- Journal of RNA and Genomics (2010) Research Article
Cell-specific RNA interference by peptide-inhibited-peptidaseactivated siRNAs
| Sandra Koehn1, Hendrik W Schaefer1, Mirko Ludwig1, Natja Haag1, Ulrich S Schubert2, Lydia Seyfarth1, Diana Imhof3, Udo R Markert1, Tobias G Poehlmann1,2* 1Placenta-Lab, Dept of Obstetrics, Friedrich-Schiller-University Jena, Bachstr 18, 07743 Jena, Germany 2Laboratory of Organic and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldstr 10, 07743 Jena, Germany 3Institute of Biochemistry and Biophysics, Friedrich-Schiller-University Jena, Hans-Knöll-Str 2, 07745 Jena, Germany |
| Corresponding Author: Tobias Poehlmann, Email: tobias.poehlmann@uni-jena.de, Tel: +49 3641 930850, Fax: +49 3641 930852 |
| Received 18 October 2010; Revised 12 December 2010; Accepted 16 December 2010; Published online 31 December 2010 |
| © Copyright The Authors: This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/). This license permits noncommercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Abstract
The use of chemically-synthesized short interfering RNAs (siRNAs) is the key method of choice to manipulate gene expression in mammalian cell cultures and in vivo. Several previous studies have aimed at inducing cell-specific RNA interference (RNAi) in order to use siRNA molecules as therapeutic reagents. Here, we used peptide-inhibited siRNAs that were activated after cleavage by cell-specific peptidases. We show that siRNAs with bound peptide at the antisense strand could be activated in target cells and were able to induce RNAi in a cell-specific manner. Green Fluorescent Protein (GFP) and Signal Transducer and Activator of Transcription (STAT)-3 gene expression were selectively reduced in a JEG-3 human choriocarcinoma cell line expressing the activating enzyme caspase-4, whereas the effect was absent in HEK cells which lacked the enzyme. In JEG-3 cells, reduction of STAT3 gene expression by conventional and peptide-inhibited siRNA led to a decrease in cell proliferation. This suggests that peptide-inhibited siRNAs provide improved cell specificity and offers new opportunities for their therapeutic use.
KEYWORDS |
| Caspase-4, peptidase, cell-specific siRNA, RNAi |
INTRODUCTION |
| RNAi-based strategies are among the leading methodologies to silence gene expression in mammalian cells. Typically, 21-bp dsRNA molecules, termed short interfering RNAs (siRNAs), with perfect complementarities in one strand to the target mRNA, are used to induce RNAi (Elbashir et al, 2001). Upon delivery into the cell, siRNAs are incorporated into the RNAinduced silencing complex (RISC) containing the signature component of the RNAi machinery, Argonaute 2 (Ago2) (Hammond et al, 2001; Martinez et al, 2002; Maniataki and Mourelatos, 2005). The siRNA strand containing the thermodynamically less stable 5’end, the antisense strand (AS), is preferentially incorporated as the guiding strand of RISC (Khvorova et al, 2003; Schwarz et al, 2003), while the sense strand (SS) of the siRNA duplex is cleaved by Ago2 and liberated (Matranga et al, 2005; Rand et al, 2005; Leuschner et al, 2006). This generates the activated RISC containing the guiding antisense strand, which binds the complementary target mRNAs and leads to their cleavage by Ago2 (Liu et al, 2004). Whereas unmodified siRNAs are highly efficient in cultured cells, chemical modification is generally considered to be a prerequisite for fulfilling the potential of siRNAs in vivo: Successful experiments in animal models have frequently relied upon injection of chemically-modified siRNAs (Lorenz et al, 2004; Soutschek et al, 2004; Mack et al, 2005). Modifications of siRNAs mostly aim at achieving better intracellular delivery (Soutschek et al, 2004) or enhanced nuclease resistance (Mack et al, 2005). Some chemical modifications of siRNAs include conjugation to lipids (Soutschek et al, 2004; Lorenz et al, 2004) and membrane-penetrating peptides (Muratovska and Eccles, 2004), or RNA aptamer sequences (McNamara et al, 2006). These modified siRNAs with superior potency require lower doses for gene silencing (Allerson et al, 2005; Elmen et al, 2005; Schulz et al, 2009), and specific chemical modifications reduce siRNA side-effects, such as the induction of recipient immune responses and inherent off-target effects (Vollmer et al, 2004). |
| In general, in vivo siRNA delivery is difficult. Another hurdle for its pharmaceutical use is to deliver siRNA exclusively into the target tissue. Known strategies for cell- or tissue-specific siRNA delivery, e.g., using receptor-ligand systems or aptamer structures, also lead to the induction of RNAi in other cells by non-specific siRNA uptake, though on a lower level (Yutaka and Kazunari, 2006). Here, we demonstrate the proof-of-principle for a mechanism to provide a key-lock-approach to siRNA molecules in order to induce RNAi specifically in the target cells. |
MATERIALS AND METHODS |
| Peptide synthesis |
| The peptide Fmoc-LE(OFm)VD(OFm)G-OH was synthesized manually by a combination of solution and solid-phase synthesis. The dipeptides H-E(OFm)V-OH and H-D(OFm)G-OH were prepared using a chlorotrityl chloride resin (500mg). The Asp and Glu derivatives were used in the Boc-Na-protected version (Bachem, Bubendorf, Switzerland), while all other amino acid derivatives were used in the Fmoc-protected form. Coupling reactions were performed using the amino acids (4eq) activated with HBTU (4eq) and DIEA (8eq) in DMF for 0.5-1hr (double couplings). Fmoc removal during solid-phase synthesis was effected by treating the resin twice with 20% (v/v) piperidine in DMF for 5min and 15min, respectively. Cleavage of the Boc-protection was accomplished concomitantly with peptide removal from the resin using TFA/water/triisopropylsilane (95:2.5:2.5) for 3hr at room temperature. The crude peptides were precipitated in diethyl ether, centrifuged and washed three times with diethyl ether. The yields of crude dipeptides were between 70-80%. Following introduction of Fmoc- Leu-F in H-E(OFm)V-OH according to a procedure described earlier (Müller et al, 1999; Ananda et al, 2000), the resulting tripeptide was coupled to H-D(OFm)G-OH in solution using TFFH as coupling reagent as reported (Carpino and El-Fahan, 1995). The crude pentapeptide Fmoc-LE(OFm)VD(OFm)G-OH was purified by reversedphase HPLC on a Shimadzu LC-10AT chromatograph (Duisburg, Germany) with a Vydac 218TP column (5μm particle size, 300Å pore size, 4.6x25mm). Peptides were eluted with a gradient of 20-90% eluent A and B within 70min at a flow rate of 1.0ml/min, where A was 0.1% (v/v) TFA in water and B 0.1% (v/v) TFA in acetonitrile; detection was at 220nm. The molar masses of all peptides were determined by MALDI-TOF mass spectrometry on a Laser Tec Research mass spectrometer (Perseptive Biosystems, Weiterstadt, Germany) using cyano-4- hydroxycinnamic acid as matrix. |
| siRNA-peptide conjugation |
| The described pentapeptide carrying protecting Fmocgroups was dissolved in dioxane and activated by pentapeptide-NHS-ester with equimolar amounts of NHS and DCC. |
| To produce PI-siRNA, the siRNA five prime end antisense strand was modified with an aminohexyl group (Eurogentec, Belgium). Because of intrinsic peptide chemistry and selection of the protecting Fmoc-groups, the peptide will only bind to the linker structure on the siRNA. For conjugation, a quintuple excess of pentapeptid-NHSester (dissolved in dioxane) was mixed with one amount of modified siRNA single strand at room temperature for 1.5hr. After lyophilization, the protecting Fmoc-groups were cleaved by adding DMF and diethylamine, the resulting PI-siRNA was dialyzed and lyophilized again. |
| siRNA and PI-siRNA single strands were separately diluted with RNAse free water and annealed by incubation at 95oC for 3min followed by 37oC for 1hr in annealing buffer (Eurogentec, Belgium). |
| Cell lines |
| JEG-3 is an adherent human placental choriocarcinoma cell line, which maintains several trophoblast-like capacities including production of pregnancy related hormones. This cell line was purchased from ATCC (USA, ATCC No: HTB-36™). HEK is a standard adherent human embryonic kidney cell line, purchased from ATCC (USA, ATCC No: CRL-1573™). For GFP-siRNA experiments, both cell lines were transfected with GFPplasmid (Clontech, USA) using electroporation (1150V, 10ms, 2 pulses) (Microporator MP-100, Peqlab, Germany) and selected by using neomycine resistance (Sigma, USA). |
| Cell culture |
| All cultures were commenced at 106 cells/175cm2 flask, and maintained under standardized cell culture conditions (37oC, 5%, v/v, CO2, humidified atmosphere) in DMEM medium (Dulbecco’s Modified Eagle Medium; Cambrex, U.K.) containing L-glutamine (Gibco, USA) supplemented with 10% (v/v) fetal calf serum (FCS; Gibco, USA) and 2% (w/v) antimycotic-antibiotic solution (AAS, Sigma, Germany). Cells were trypsinized when confluence was estimated to be over 75%. |
| Analysis of caspase-4 activity |
| A total of 106 cells were cultured in 6-well plates at 50% confluency. Medium was removed and 1ml fresh medium was given into each well. 20μl of caspase-4 substrate (LE- V-D-G-AFC, MoBiTec, Germany, 20mM) was added to the medium. After 4hr of incubation at standard conditions, medium was removed; cells were washed with 1ml HBSS and lysed in 100μl of lysis buffer (20mM HEPES, 2mM EGTA, 0.2mM EDTA, 0.12M NaCl, 1%, v/v, Triton-X 100) supplemented with protease inhibitors (5mM beta-glycero- phosphate, 3mM MgCl2, 20mg/ml aprotinine (trasylol), 20mg/ml leupeptine, 5mg/ml pepstatine A, 647ng/ml anti-pain, 10mg/ml bestatine, 0.1mM PMSF, 1mM sodium orthovanadate). Afterwards, cell lysates were analyzed for cleaved fluorometric substrate by using HPLC and fluorescence detector as described by Koehn et al (2008). |
| RNA interference |
| Oligonucleotides were designed to interfere exclusively with GFP mRNA (Eurogentec, Belgium) or STAT3 mRNA (sequence: 5’AAT GTT CTC TAT CAG CAC AAT) as described elsewhere (Poehlmann et al, 2005). As a negative control, the same nucleotides were scrambled to form a non-genomic sequence (Sequence: 5’AAG CCA CTT ATA AAT TCG TTC; Eurogentec, Belgium). In the previous studies, the used siRNA sequences showed efficient silencing rates and no off-target effects (Poehlmann et al, 2005). During electroporation, 21- nucleotide siRNA and PI-siRNA duplexes were applied at a concentration of 1μM (in electroporation pipette; final concentration in cell culture was 10nM); siRNA delivery using RiboJuice transfection reagent (Merck, Germany) was performed at a final concentration of 5nM. For electroporation, cells were trypsinized and washed with HBSS. After centrifugation, cell pellets were resuspended in 100μl cell resuspension buffer (Peqlab, Germany). 1μl of control, target-gene siRNA or PI-siRNA solution (100μM) was added to cells and electroporation was performed in 2 pulses at 1100V for 35ms (Microporator MP-100). After the electroporation procedure, cells were cultured in 6-well plates with 2ml DMEM medium per well. For siRNA delivery using RiboJuice transfection reagent, cells were cultured in 6-well plates in standard DMEM medium. At 70% confluency, cells were washed with PBS and 1ml new DMEM medium was added. A volume of 2μl RiboJuice was added to peptide-inhibited or conventional siRNA (1μM) and incubated for 20min at room temperature. Afterwards samples were applied to cells. Analyses were performed 24-96hr after transfection. |
| Caspase-4 inhibition |
| JEG-3 cells were cultured under standard conditions and prepared for siRNA transfection as described above. Caspase-4 inhibitor (Z-YVAD-FMK; R&D Systems, USA) was applied to JEG-3 cells during siRNA transfection at a final concentration of 200nM. |
| Flow cytometry |
| GFP transfection efficiency and RNAi were analysed using flow cytometry. Cells were cultured in 6-well plates, washed with PBS and trypsinized. DMEM-medium supplemented with 10% (v/v) fetal calf serum (FCS; Gibco, USA) and 2% (w/v) antimycotic-antibiotic solution (AAS, Sigma, Germany) was added to stop trypsin activity. Fluorescence intensity and the number of GFP positive cells were assessed using a FACSCalibur (BD, USA) and Cellquest Pro software (BD, USA). |
| Polyacrylamide gel electrophoresis (PAGE) and western blot |
| A number of 2x106 cells were cultured in 6-well plates, washed with PBS containing proteinase inhibitors and scraped from the culture plates. After centrifugation, cell pellets were suspended in 100μl lysis buffer supplemented with protease inhibitors. Protein concentrations of cell lysates were determined using a kit based on the Bradford method (Sigma, USA). 20mg of whole cell extract was solublized in gel loading buffer (62.5mM Tris/HCl pH 6.8; 2%, w/v, SDS; 25%, v/v, glycerol; 1% (w/v) phenol blue; 5% (v/v) beta-mercaptoethanol), denatured for 5min and separated on 10% (w/v) acrylamide-SDS gels (Serva, Germany). After protein transfer, nitrocellulose membranes were blocked in NET-G buffer (150mM NaCl, 50mM Tris-HC1, 5mM EDTA, 0.25%, w/v, gelatin;. pH 7.4) for 1hr. Antibodies against STAT3 (Cell signaling, USA) were applied for 12hr at a 1:1000 dilution. For detection, peroxidase-conjugated anti-rabbit IgG (New England Biolabs, USA) was employed at a dilution of 1:10,000 for 1hr. Visualisation was performed using a LumiGlo Chemiluminescence reagent (New England Biolabs, USA). Protein loading control of western blots was performed by application of anti-tubulin antibodies (Sigma, USA) following the same detection procedure as described for STAT3. All western blots were repeated 3 times and were analyzed and quantified by Totallab TL100 software, version 2008. |
| Proliferation assay |
| 24hr after transfection, cells were trypsinized and counted by a flow cytometry method (Beckman Coulter, USA). Briefly, 20,000 cells were plated in 12-well plates and incubated for 48hr. Then cells were trypsinized and counted again. Proliferation index (PI) was calculated by the following formula: |
| PI = log2(cell number/20.000) |
| The assays were done in duplicates and repeated twice. |
| Statistics |
| To investigate the significance of the results a paired t-test was performed. Differences between samples were defined as significant, for p<0.05; p<0.01 were defined as highly significant. |
RESULTS |
| Experimental design |
| We generated siRNA that induces RNAi specifically in target cells. The enabling mechanism to induce specificity relies on the activation of a peptide-inhibited siRNA (PIsiRNA) by cell-specific active peptidases (Figure 1). Since binding of sterically large structures (e.g., peptides) to the backbone of the siRNA antisense strand reduces the induction of RNAi (Chiu and Rana, 2003), a complete inhibition of the siRNA by an oligopeptide was attempted. After peptide cleavage a small chemical group should remain on the siRNA in order to minimize its influence on the siRNA activity. Therefore, an aminohexyl linker was used to conjugate the specific target peptide for caspase-4 with the siRNA (Figure 2a) and the very same peptide sequence was used to analyse caspase-4 activity. After peptide cleavage, the aminohexyl linker and the glycine residue should remain attached to the siRNA (Figure 2b). In non-target cells (Figure 1) the peptide-siRNA-chimera remains stable without inducing RNAi to a significant level. |
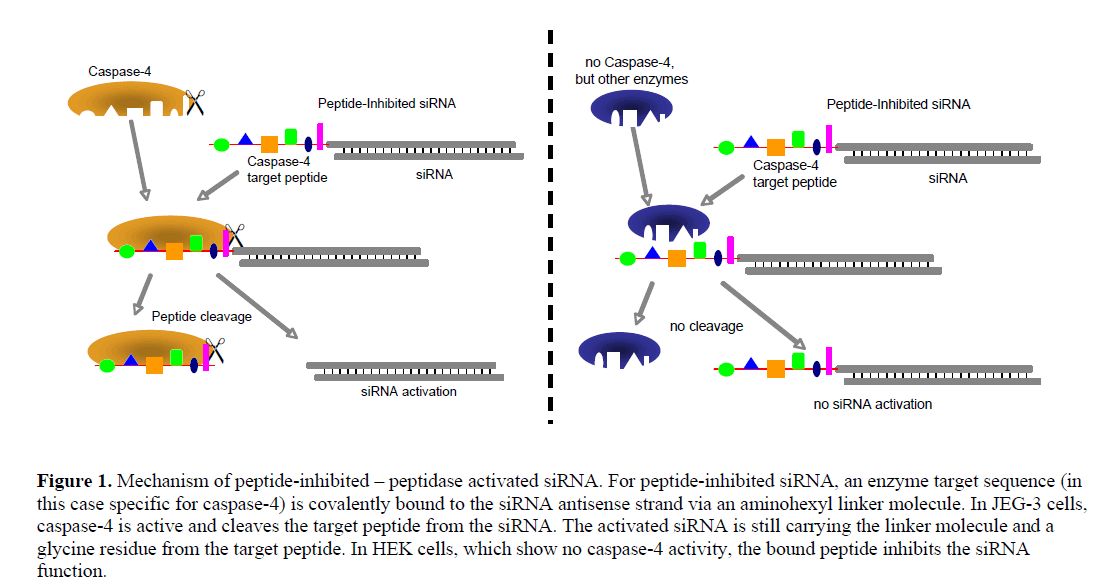 |
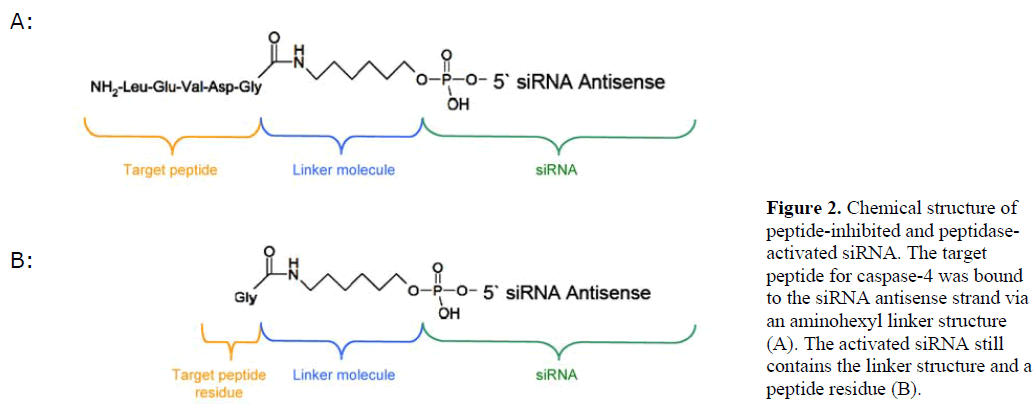 |
| In order to test the described concept, the two cell lines JEG-3 and HEK were used because of their similar characteristics regarding siRNA transfection and their presumed difference in caspase-4 activity. The differentially-expressed enzyme is well suited for being used with the PI-siRNA strategy as it cleaves its target peptide L-E-V-D-G between aspartic acid and glycine, leaving only the small glycine residue at the siRNA linker after peptide cleavage. We tested the proteolytic activity of caspase-4 and produced two PI-siRNAs. GFP and STAT3 were chosen as target genes and the caspase-4 target sequence was chosen as bound peptide. After the protein expression analyses, proliferation of JEG-3 cell including subsequent transfection of PI-siRNAs was analysed. Additional time dependent STAT3 expression analysis was performed in order to investigate the level of PIsiRNA activation and to compare its induction kinetics with that of conventional siRNA. Furthermore, a rescue experiment inhibiting Caspase-4 in JEG-3 cell would support the enzymatic activation of the PI-siRNA concept. |
| PI-siRNA synthesis |
| An aminohexyl linker was chosen for conjugation to obtain a stable bond between the peptide and the siRNA antisense strand. The coupling chemistry used aimed at, (i) high binding efficacy of the peptide to the siRNA in order to prevent unspecific siRNA effects, and, (ii) siRNA stability in order to prevent siRNA degradation. The predicted chemical structure of the desired PI-siRNA was analysed by MALDI-TOF mass spectrometry and gel electrophoreses independently confirmed the high coupling rate (data not shown). |
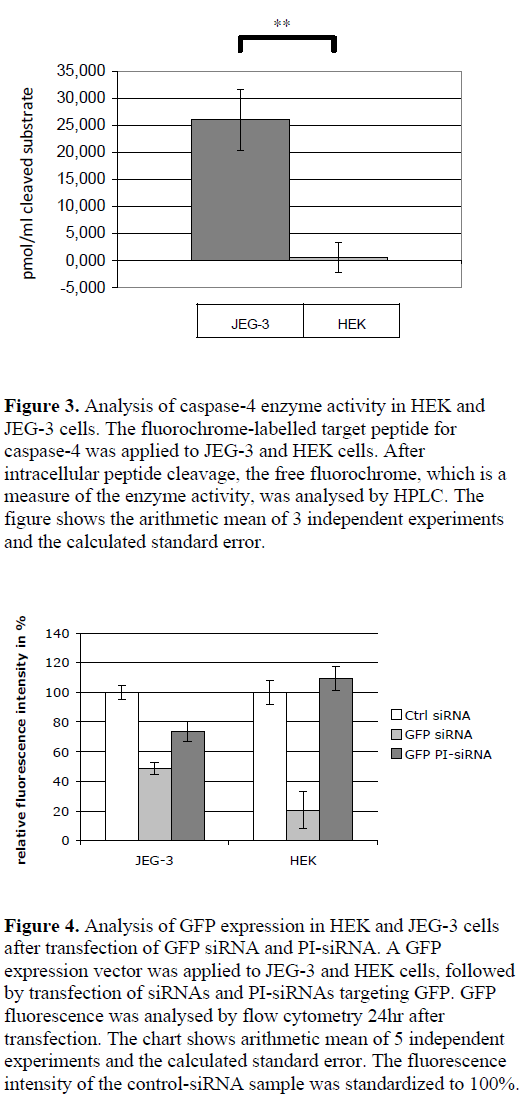
| Caspase-4 activity in JEG-3 and HEK cells |
| By using a synthetic, fluorochrome-labelled caspase-4 substrate, the peptidase activity in the cytoplasm of JEG-3 and HEK cells was analysed via HPLC by measuring the free fluorochrome after cleavage. JEG-3 cell lysates showed a measurable enzyme activity (cleavage of 26 pmol/ml cleaved substrate in the cell lysates), whereas no cleavage of caspase-4 substrate was detected in HEK cells (Figure 3). |
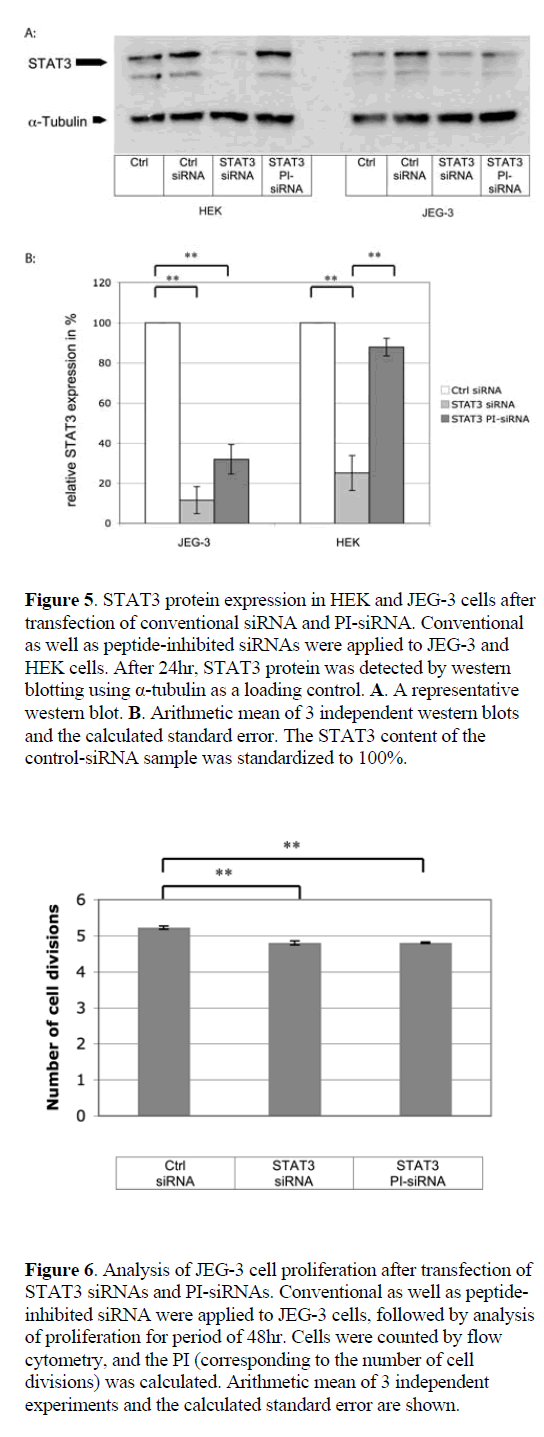
| Cell-specific gene silencing by peptidase-activated siRNAs |
| Conventional siRNAs reduced GFP expression by 51.4% in JEG-3 and by 79.3% in HEK cells, as compared to nongenomic scrambled control siRNA (Figure 4). PI-siRNA molecules induced a GFP silencing efficacy of 26.6% in JEG-3 cells, while no silencing was detected in the caspase- 4-negative HEK cells. As anticipated, GFP siRNA silencing was exclusively induced in caspase-4 positive JEG-3 cells, although the RNAi effect was weaker than expected. |
| Similarly, STAT3 PI-siRNA knock-down was specific to JEG-3 cells. Figure 5a shows the western blot analysis indicating the cell-specific silencing effect of PI-siRNA. As shown by the arithmetic mean of three independent western blots (Figure 5b), conventional siRNAs were able to reduce STAT3 expression by around 90% in JEG-3 and by 75% in HEK cells; whereas PI-siRNA decreased STAT3 expression in JEG-3 cells by 70% and 13% in HEK cells. |
| Analysis of JEG-3 cell proliferation after STAT3 silencing |
| We aimed at analyzing the modulation of cell proliferation after STAT3 silencing induced by conventional STAT3 siRNAs and PI-siRNAs. Our results indicate that cells treated with conventional and PI-siRNAs showed a similar reduction of cell proliferation (PI=4.8) as compared to those transfected with control siRNA (PI=5.2) (Figure 6). |
| Analysis of activation kinetics of peptidase-activated siRNAs |
| To exclude an influence of the transfection method on the activity data for peptide-inhibited siRNA, a chemical siRNA delivery agent was used. In general the same effects were obtained as for the electroporation approach. To investigate the kinetics of peptidase activation, we analysed STAT3 silencing rates 48hr, 72hr and 96hr post-transfection (Figure 7). In JEG-3 cells conventional siRNA was active during the whole period of 96hr, whereas in HEK cells STAT3 expression returns to the normal level at 96hr. Using a chemical delivery method both, peptide inhibited and conventional siRNA, display maximal efficacy at 72hr in JEG-3 target cells. In contrast to conventional siRNA, the gene expression becomes elevated again after 96hr in JEG-3 cells. In non-target HEK cells PI-siRNAs remained inactive and did not induce detectable RNAi. |
| Specific inhibition of Caspase-4 in JEG-3 cells |
| In order to investigate whether the silencing effect in JEG- 3 cells was due to PI-siRNA activation by capase-4 or due to unspecific peptide degradation, a caspase-4 inhibitor was delivered to JEG-3 cells together with PI-siRNAs. By inhibiting the caspase-4 pathway, only conventional siRNAs reduced STAT3 expression, whereas PI-siRNAs was unable to induce RNAi (Figure 8). |
 |
DISCUSSION |
| Peptide-inhibited-peptidase-activated siRNAs exhibit a new mechanistic alternative to obtain cell-specific activation of siRNA molecules in target cells (Figure 1). The challenge of this strategy is to find a target peptidase with cell-specific activity, which is able to activate the siRNAs by peptide cleavage. Therefore, a dual cell line experiment (JEG-3 and HEK cells) using caspase-4 as peptidase was performed to show the feasibility of the concept. The system was chosen because of the enzyme properties (cleavage site at the end of the target peptide) and the fact that caspase-4 is expressed in JEG-3 cells (Fitzgerald et al, 2005), but according to microarray data does not show significant expression in kidney cells. After peptide cleavage, a remaining glycine residue and an aminohexyl linker are left attached to the siRNA antisense strand (Figure 2b). The full caspase-4 target pentapeptide should prevent gene silencing since the five prime terminus of guide strands bind to PIWI domain of AGO2 protein, which is critical to effective RNAi. To validate the hypothesis, we first analyzed the activity of caspase-4 in target and non-target cells. Subsequently, we produced a peptide-inhibited siRNA carrying caspase-4 substrate at the 5’end of the antisense strand and analysed its chemical structure before testing the cell-specific biological activity of the described peptide-inhibited siRNA targeting GFP as well as STAT3 expression. Finally, we investigated the influence of PI-siRNA targeting STAT3 on the proliferation of JEG-3 target cells. |
 |
| Peptide-siRNA coupling |
| A general challenge is to find a chemical strategy for efficiently coupling a peptide to an siRNA without degrading the siRNA. The F-moc strategy applied to protect side groups of the peptide lead to correct coupling to the aminohexyl-linker of the siRNA as analysed by MALDI-TOF mass spectrometry. Gel electrophoresis showed high coupling efficacy of the chemical strategy. |
| Various cell types show different caspase-4 activity |
| Caspase-4 is a cysteine protease and is believed to be an inflammatory caspase, along with caspase-1 and caspase-5 (Yi et al, 2009). Presumably, caspase-4 is not involved in apoptotic processes. Its expression is increased in highly invasive and proliferatives choriocarcinoma and embryonic trophoblast cells compared to lower invasive control cell types (Fitzgerald et al, 2005). Even though caspase-4 is expressed in multiple cancer cells, it is not clear whether or not a correlation exists between prevention of apotosis in cancer cells, cancer development and caspase-4 expression and/or activity (Xie et al, 2008; Jiang et al, 2009; Kim et al, 2009). |
| In the present work, we investigated whether the cellspecific activity of caspase-4 could be exploited for activation of siRNA inhibited by a target peptide for caspase-4 bound covalently to the siRNA antisense strand. In our experiments we used two different cell lines: JEG-3 cells displaying high caspase-4 activity, and HEK cells with nearly inactive caspase-4. Despite the claim that commercially available caspase substrates are not necessarily cleaved by the respective enzyme (Berger et al, 2006), we showed a cell-specific cleavage of the substrate used. It can thus be concluded that PI-siRNA carrying the same peptide should be cleaved in a cell-specific manner. |
| siRNA-peptide-chimera induce cell-specific RNAi |
| Most strategies to induce cell specific RNAi apply siRNA in the form of a suitable non-covalently bound complex since, after delivery, these structures will release an unmodified siRNA (Simeoni et al, 2003; Crombez et al, 2007; Krajcik et al, 2008). Covalently-bound siRNA chimera contained, however, unstable disulfide-bounds, which were cleaved after delivery to the cells (Detzer et al, 2009). Stable modifications of the siRNA are in most cases confined to the sense strand of the siRNA and are rather small to avoid any influence on RISC complex formation. Our data indicate that a sequence of five amino acids covalently bound to the antisense strand of a siRNA is sufficient to inhibit its biological activity. This is confirmed by findings showing that the biological activity of 18-21bp siRNA exceeds that of larger RNA molecules (Elbashir et al, 2001; Scherr et al, 2003). As expected, the peptide-inhibited siRNA did not show activity in HEK cells, which do not express the specific peptidase (caspase- 4) to cleave the peptide from the siRNA. In JEG-3 cells exhibiting caspase-4 activity the PI-siRNAs were able to silence gene expression, indicating that the peptide was cleaved from the siRNA. Interestingly, 48hr after delivery the PI-siRNAs did not reach the same biological activity as conventional siRNA. |
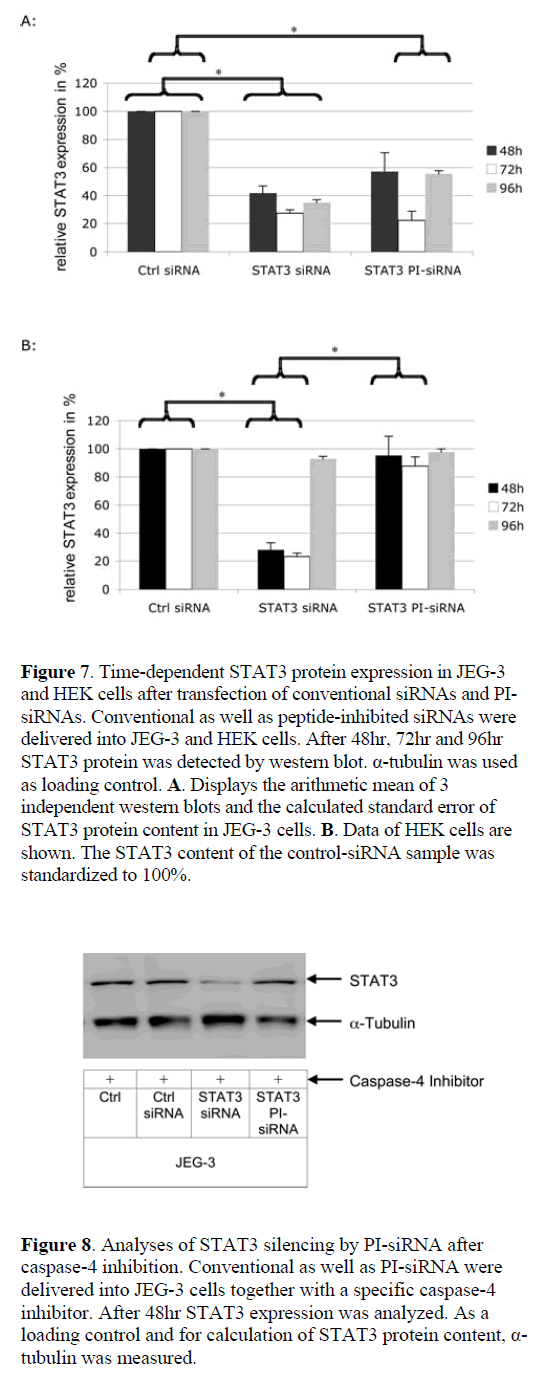 |
| To validate that this is a time-dependent effect due to the fact that the peptide has first to be cleaved from the siRNA before RNAi and not an artifact induced by the Glycin residue on the linker or the linker structure itself, gene expression was measured after 48hr, 72hr and 96hr. Moreover, an insufficient PI-siRNA delivery into nontarget cells could result in an apparent cell-specific effect in non-target cells. Therefore, a chemical delivery strategy was used instead of electroporation. These experiments had four major results: (i) PI- siRNAs remain stable and inactive in non-target HEK cells; (ii) PI-siRNAs show their maximal silencing effect 72hr after delivery, which is slower than conventional siRNAs and is expected due to the time consuming cleavage process; (iii) PI-siRNAs and conventional siRNAs display the same silencing effect, and; (iv) the cell-specific effect of PI-siRNA was not due to insufficient delivery. |
| The finding that the induction of RNAi by PI-siRNA occurs at a later time point than by conventional siRNA is supported by recent data that suggest a maximal cleavage capacity of caspase-4 around 4-10hr after contact with its target peptide (Koehn et al, 2008). |
| Since the antisense 5’phosphate group binds to RISC it will be topic of further investigations how a siRNA carrying a glycine and a linker structure at the 5’end is incorporated into RISC and is able to induce RNAi. |
| Differential gene silencing of STAT3 restricts tumor growth in vitro |
| In previous work it was shown that STAT3 is a crucial regulator of embryonal trophoblast invasion during human pregnancy and that its constitutive activation is often correlated with extensive tumor invasion and poor therapeutic outcome (Rebouissou et al, 2009; Ren et al, 2009; Thoennissen et al, 2009). In earlier studies it was found that silencing of STAT3 in JEG-3 choriocarcinoma cells reduces their invasive capacity and was not correlated with the induction of apoptosis (Poehlmann et al, 2005). In the present study, proliferative properties of JEG-3 cells were reduced when STAT3 expression was silenced. This effect can be induced, in an analogous manner, both by conventional siRNAs and by PI-siRNAs. Since proliferation was measured 72hr after siRNA delivery, these findings additionally support that the efficacy of PIsiRNAs and conventional siRNA was comparable. |
| PI-siRNA controls |
| In the presented work for all experiments a negative siRNA control sequence and a positive GFP or STAT3 siRNA sequence was used. The authors acknowledge that a second PI-siRNA control targeting GFP or STAT3 expression that is not cleaved in any of the used cell lines would be useful. The challenge in this regard was to find a peptide sequence that was not cleaved by any other cytoplasmatic peptidase of the cells other than caspase-4 and at the same time has the same charge and size as the caspase-4 target peptide applied. |
| To validate if the cell specific gene silencing by PIsiRNAs was due to caspase-4 activation, a caspase-4 inhibitor was delivered into JEG-3 cells together with PI-siRNAs. This way it was clearly shown, that the effects of the presented PI-siRNA were caspase-4 dependent. |
CONCLUSIONS |
| The present work provides a model for targeted, cellspecific activation of peptide-inhibited siRNAs. This new tool might be useful for RNAi applications in co-cultures in vitro, but further development may also lead to applications in vivo. Combined with established technologies of target cell-specific siRNA delivery, this technology might reduce side-effects of siRNAs in nontarget cells and lead to increased cell-specificity in therapeutic applications. |
ACKNOWLEDGMENTS |
| This work was supported by a grant of the Ministry of Economics and Technology (FKZ 03EFT2TH04), the Stiftung für Technologie, Innovation und Forschung Thüringen, STIFT and the Friedrich-Schiller-University Jena, Germany. We are grateful to Anja Baumgärtel for her support during mass spectrometry measurements and to Professor Dr Dieter Schubert, Professor Dagmar Fischer, Professor Achim Aigner and Dr Rolf Günther for their critical reading of the manuscript. |
COMPETING INTERESTS |
| None declared. |
LIST OF ABBREVIATIONS |
| DCC; N,N'-Dicyclohexylcarbodiimide |
| DIEA; N,N-diisopropylethylamine |
| DMF; (N, N-Dimethylformamide) |
| Fmoc-Leu-F; aminoacid Leucine containing protecting fmoc groups |
| HBSS; Hank's Buffered Salt Solution |
| HBTU; (2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) |
| H-D(OFm)G-OH; peptide DG containing protecting fmoc groups |
| H-E(OFm)V-OH ; peptide EV containing protecting fmoc groups |
| NHS; N-Hydroxysuccinimid |
| PBS; Phosphate buffered saline |
| PI-siRNA; peptide-inhibited-peptidase-activated siRNA |
| TFA; trifluoroacetic acid |
| TFFH; Tetramethylfluoroformamidinium hexafluorophosphate |
References |
|