- Journal of RNA and Genomics (2005) Review Article
A novel approach for inhibition of HIV-1 by RNA interference: counteracting viral escape with a second generation of siRNAs
| Olivier ter Brake and Ben Berkhout* Department of Human Retrovirology, University of Amsterdam, Academic Medical Center (AMC), Meibergdreef 15, 1105 AZ Amsterdam, The Netherlands |
| *Correspondence to: Ben Berkhout, Email: b.berkhout@amc.uva.nl, Tel: +31 20 5664853, Fax: +31 20 6916531 |
| Journal of RNAi and Gene Silencing (2005), 1(2), 56-65 |
| © Copyright Ter Brake and Berkhout |
| (Received 02 September 2005; Revised 06 October 2005; Accepted 06 October 2005, Available online 14 October 2005; Published 14 October 2005) |
Abstract
RNA interference (RNAi) is an evolutionary conserved gene silencing mechanism in which small interfering RNA (siRNA) mediates the sequence specific degradation of mRNA. The recent discovery that exogenously delivered siRNA can trigger RNAi in mammalian cells raises the possibility to use this technology as a therapeutic tool against pathogenic viruses. Indeed, it has been shown that siRNAs can be used effectively to inhibit virus replication. The focus of this review is on RNA interference strategies against HIV-1 and how this new technology may be developed into a new successful therapy. One of the hallmarks of RNAi, its sequence specificity, also presents a way out for the virus, as single nucleotide substitutions in the target region can abolish the suppression. Strategies to prevent the emergence of resistant viruses have been suggested and involve the targeting of conserved sequences and the simultaneous use of multiple siRNAs, similar to current highly active antiretroviral therapy. We present an additional strategy aimed at preventing viral escape by using a second generation of siRNAs that recognize the mutated target sites.
Keywords |
| HIV-1, RNA interference, gene therapy, siRNA, shRNA, combinatorial therapy, lentiviral vector |
Introduction |
| Highly-active antiretroviral therapy (HAART), which involves the use of multiple antiviral agents, has been very successful in reducing the rates of progression to AIDS and death in people with HIV-1. However, adverse effects of the long-term use of these drug regimens, like toxicity and the emergence of drug resistant variants, obviate the need for new and improved methods to suppress HIV-1 in infected individuals. Recently, it was shown that delivery of siRNAs in mammalian cells can trigger a highly specific RNA interference response resulting in knockdown of the gene of interest. This finding raised the possibility that RNA interference can be used as a potent antiviral tool against HIV-1. |
| siRNAs targeting HIV-1 sequences can inhibit virus production and virus replication in transient transfection experiments (Capodici et al, 2002; Coburn and Cullen, 2002; Hu et al, 2002; Jacque et al, 2002; Lee et al, 2003; Park et al, 2003; Dave and Pomerantz, 2004). The transient nature of these methods is not desirable in a therapeutic setting dealing with a persistent viral infection. Although patients may be treated with a stabilized form of siRNAs, the risk of acquiring resistant HIV-1 mutants is relatively high because the concentration of siRNAs will vary in different body compartments. Furthermore, delivery and toxicity remain major issues for a treatment with synthetic siRNAs. In contrast, if patients are treated with a stably integrated viral vector expressing a steady supply of interfering RNAs, HIV-1 replication will be inhibited constantly. siRNAs can be expressed from polymerase-III transcription units. The sense and anti-sense strands can be expressed from two different promoters, and RNAi is triggered upon annealing of the two strands (Lee et al, 2002; Banerjea et al, 2003; Tran et al, 2003). More potent inhibition was achieved when the sense and antisense strand were expressed as a single transcript with the ability to from a duplex hairpin structure (Brummelkamp et al, 2002). HIV-1 replication can be effectively inhibited with such short hairpin RNAs (shRNA) in stably transduced cell lines (Boden et al, 2003a; Unwalla et al, 2004; Das et al, 2004; Boden et al, 2004a; Lee et al, 2005). |
| Because RNAi is highly sequence specific, a single point mutation can potentially result in full resistance to RNAi. Indeed, resistant variants can emerge when a single antiviral shRNA is used (Boden et al, 2003a; Das et al, 2004). The emergence of RNAi-resistant mutants poses a challenge for the development of RNAi based treatments for HIV-1 infected individuals. It has been suggested to use multiple highly-effective shRNAs that target conserved regions of the viral genome (Berkhout, 2004). More potent suppression will further reduce the ability of the virus to evolve, and escape from multiple shRNA inhibitors will require several mutations. In this report, we will present an overview of current publications on RNAi against HIV-1. Furthermore, we have analyzed all published HIV-1 targets and present an outline for the effective use of RNAi against HIV. This includes a new strategy to prevent the emergence of resistant variants by including additional shRNAs that are directed against the most likely escape mutants. |
| HIV-1 escapes from a single shRNA |
| Two recent studies addressed the potency and durability of anti-HIV RNAi approaches. A short hairpin RNA (shRNA) against the tat gene was expressed from an H1- promoter in an adeno-associated virus vector (Boden et al, 2003a). Potent inhibition of HIV-1 replication was scored, but an escape virus appeared in prolonged cultures. Similar results were described for a shRNA against the Nef gene using a retroviral vector (Das et al, 2004). The latter study described 7 independent HIV-1 escape variants. These studies convincingly demonstrate that inhibition is potent and sequence-specific, but also that HIV-1 is able to escape from the inhibitory action of a single shRNA. Boden et al described a single escape virus with a point mutation in the target sequence, and Das et al (2004) described a multitude of escape routes (point mutation, double point mutation, partial or complete deletion of the target sequence). The possibility of virus escape seems even more apparent with the recent demonstration that HIV-1 can also gain resistance by a point mutation outside the target sequence (Westerhout et al, 2005). This mutation was demonstrated to change local RNA folding, such that the target sequence becomes inaccessible to the RNAi machinery. The emergence of resistant virus variants poses a serious problem for using RNA interference in a therapeutic setting. |
| Counteracting viral escape with multiple shRNAs |
| One strategy to counteract viral escape is to use multiple shRNAs targeting different conserved regions of HIV-1 (Haasnoot et al, 2003; Stevenson, 2003; Hannon and Rossi, 2004; Berkhout, 2004; Shankar et al, 2005). Conserved targets in the HIV-1 RNA genome will not allow for a deletion-based escape route, as occurred with the shRNA targeting the non-essential Nef gene sequences (Das et al, 2004). One can try to estimate the chance of escape with one versus multiple shRNAs. First, we make the assumption that deletion is not an option for the virus; only point mutations are allowed to occur. In principle, a single point mutation can make the virus insensitive to RNAi. The error rate of the reverse transcriptase of HIV-1 is 3 x 10-5 (Mansky, 1996), consequently the chance of viral escape for a 19 nucleotide target in a single infection is 19 x (3 x 10-5) = 5.7 x 10-4. Studies in the field of drug resistance indicate that an untreated HIV infected individual has a virus population size of 104 to 105. This means that for each shRNA, several potential escape variants are already present before the start of therapy. Thus, the emergence of a drug resistant variant seems inevitable when a single shRNA is used. When multiple shRNAs (N) are used simultaneously, the likelihood of obtaining a drug resistant variant drops exponentially with the number of shRNAs (5.7 x 10-4)N. If we assume that there is already resistance to at least one of the shRNAs used in a patient, than the chance of a resistant variant emerging is (5.7 x 10-4)N-1. For instance, if four shRNAs are used simultaneously, the chance of escape is 1.9 x 10-10. Although this chance seems remote given the average viral load in a patient, it cannot be excluded that multi-shRNA resistant mutants can evolve through recombination. |
| That resistance can evolve, even after prolonged in vitro culturing, indicates that suppression of HIV-1 replication is not absolute when a single shRNA is used. Recently, it was shown that inhibition of a target gene is increased proportionate to the shRNA expression level. This result was obtained by combining multiple expression cassettes encoding the same shRNA in a single vector (Gonzalez et al, 2005). In addition, the combination of two different shRNAs resulted in effective and simultaneous inhibition of two targets, while their individual activity was maintained (Anderson et al, 2003; Schubert et al, 2005a). These results indicate that, when a multiple shRNA strategy would be used against HIV-1, the magnitude of inhibition will increase, perhaps approaching complete inhibition, combined with a severely reduced chance of escape. |
| Target site selection |
| During the HIV-1 life cycle, there are several steps at which RNA interference may inhibit replication (Figure 1A). Upon virus infection, the incoming RNA genome is a potential target. There is controversy with regards to targeting of the incoming HIV-1 genome with RNAi. Several publications report RNAi-mediated degradation of the RNA genome during infection in cells transfected with siRNAs (Capodici et al, 2002; Coburn and Cullen, 2002; Jacque et al, 2002), while others reported no effect at all (Surabhi and Gaynor, 2002; Hu et al, 2002). These contradicting results may perhaps be explained by differences in target accessibility in the context of the packaged RNA genome, since viral and/or cellular proteins may protect some regions, but not all. Variations in experimental conditions, e.g., the cell types used, differences in transfection efficiency, variable amounts of siRNAs or infectious HIV- 1 virus used, may also have caused these differences. Recently, it was shown that cells expressing shRNAs from an integrated lentiviral vector effectively blocked integration of the DNA provirus at low multiplicity of infection; this effect was overcome with a high dose of virus (Joshi et al, 2005). This result shows, in a relevant gene therapy setting, that the incoming HIV-1 genome can be subject to RNAimediated degradation. |
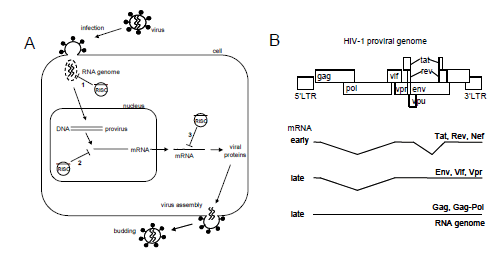 |
| Figure 1. The HIV-1 life cycle and possible opportunities for RNAi-mediated intervention. (A) After virus infection, the HIV-1 core with the RNA genome will be released into the cytoplasm. The incoming RNA genome represents a desirable target, since destruction of the genome (route 1) prevents reverse transcription and integration of the provirus. After integration of the provirus, viral messenger RNAs are expressed. Viral mRNAs can potentially be degraded in the nucleus (route 2) or in the cytoplasm (route 3), preventing viral protein production and virus particle assembly and release. (B) The proviral DNA genome. Early gene expression results in fully spliced mRNAs encoding for Tat, Rev and Nef. Late gene expression results in partially spliced mRNAs, encoding for Env, Vif, Vpr, and unspliced mRNA for Gag and Gag-Pol proteins or as the genomic RNA. |
| Late in infection, viral mRNAs are transcribed from the integrated provirus. The early viral transcripts are processed, resulting predominantly in spliced transcripts in the cytoplasm (Figure 1B). Once sufficient Rev protein is made, partially spliced messengers and unspliced genomic transcripts will be exported out of the nucleus into the cytoplasm. It is generally believed that RNA interference is a cytoplasmic phenomenon (Meister and Tuschl, 2004; Shankar et al, 2005). Since it may be beneficial to block early viral gene expression, one may prefer HIV-1 target sequences that are present in (all) early transcripts (Purcell and Martin, 1993). For instance, down-regulation of Tat and Rev protein expression may block virus production early on. In the absence of Tat protein, no induction of the viral LTR promoter occurs and without Rev the shift towards unspliced RNA cannot be made. In contrast, targeting sequences present exclusively in late unspliced mRNAs will allow for the production of early spliced mRNAs and early viral proteins. |
| It has been shown that siRNAs targeting early transcripts down-regulate all transcripts after infection with HIV-1 (Coburn and Cullen, 2002; Lee et al, 2003). Contrary to expectation, an siRNA targeting a gag sequence that is exclusively present in unspliced, late mRNA gave a similar result (Novina et al, 2002). Although this result may reflect the combined effect of the destruction of the incoming genome and the targeting of viral messengers, it may also hint at the possibility that viral transcripts are cleaved by RNAi in the nucleus prior to splicing. This option was previously discarded because RNAi was considered to be an exclusively cytoplasmic event (Novina et al, 2002). Supporting the concept of nuclear targeting of HIV-1 mRNAs, both nuclear and cytoplasmic viral RNA was downregulated by a lentiviral shRNA against the late vpu transcript (Chang et al, 2005). It was recently shown that RNA interference can effectively degrade mRNAs that are localized in the nucleus of mammalian cells (Robb et al, 2005). Similar results were obtained with shRNAs delivered by a lentiviral vector (Langlois et al, 2005). These studies suggest that RNAi may also occur in the nucleus, thus explaining the results obtained with siRNAs that target late viral transcripts. |
| In addition to the degradation of RNA, which is the focal point of this review, two other functions of RNA interference may also contribute to inhibition of HIV-1 replication. First, the RNA interference silencing complex may silence mRNA translation. This process is less sensitive to sequence variation since perfect basepairing of siRNA and mRNA is not required. Second, inhibition can also occur through transcriptional silencing mediated through chromatin remodeling (Morris et al, 2004; Kawasaki and Taira, 2005). This would prevent the expression of viral transcripts. It was recently shown that targeting of the HIV-1 LTR promoter region results in sustained inhibition (Suzuki et al, 2005). Interestingly, methylation of CpG motifs was observed in and surrounding the siRNA target regions, implicating that transcriptional silencing suppressed HIV-1 replication. |
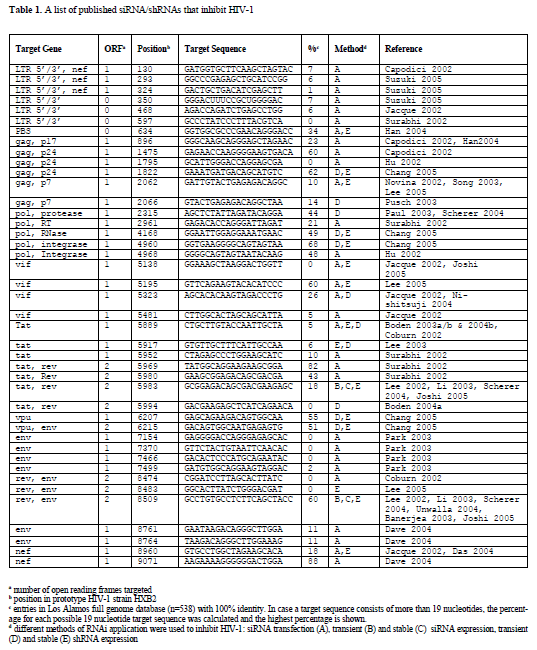
| Since there is support for shRNA induced targeting of the incoming viral RNA genome and also suggestive evidence that viral mRNAs may be degraded prior to splicing in the nucleus, we propose that target sites can be chosen throughout the HIV-1 genome. Selection can therefore be based solely on conservedness of the target sequence in different HIV-1 isolates. We have listed all 42 published siRNAs and shRNAs that were successfully used to inhibit HIV-1 production or replication (Table 1). It is important to note that each study used different ways to score inhibition of HIV-1 production, and in a few cases HIV-1 replication, such that a direct comparison is not possible. There is ample evidence that effective siRNAs against HIV-1 can be transformed into effective shRNAs (Coburn and Cullen, 2002; Jacque et al, 2002; Han et al, 2004; Das et al, 2004; Nishitsuji et al, 2004; Boden et al, 2004a; Lee et al, 2005). We aligned all 42 RNAi target sequences with 538 HIV-1 full genome sequences available from the Los Alamos National Laboratory database using the PrimaAlign tool available from their website (http://www.hiv.lanl.gov). We calculated the percentage of viral isolates that show complete similarity to the published target sequence (Table 1). Nine of the 42 target sequences are conserved in at least half of known isolates, which represent the best candidates for the multiple shRNA approach. |
| Counteracting viral escape with a 2nd generation of shRNAs |
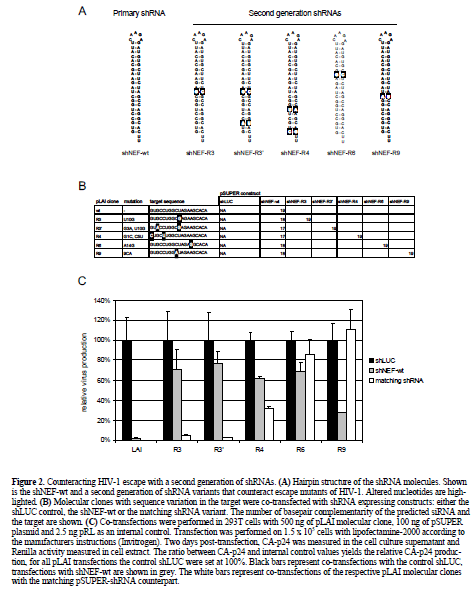
| Although, the Nef gene target (Jacque et al, 2002; Das et al, 2004) is not highly conserved (Table 1) and, therefore, not perfectly suited for an RNAi based therapy, the extensive set of viral escape variants described for this RNAi treatment (Das et al, 2004) provided us with a tool to test a different strategy to prevent viral escape. The novel approach is to include a second generation of shRNAs that target the most likely escape variants (Figure 2A). We constructed pSUPER vectors (Brummelkamp et al, 2002) expressing shRNAs directed against the mutated Nef sequences of the escape viruses (Figure 2B). HIV-1 molecular clones, based on pLAI (Peden et al, 1991), with one or two nucleotide substitutions in the target (Figure 2B), were co-transfected with a control pSUPER shLUC vector (Figure 2C, black bars) and relative virus production, as measured by Gag CA-p24 production, was set at 100%. Cotransfection of these molecular clones with the shNEF-wt plasmid showed strong inhibition of wild-type pLAI, but not of the variants with a mutated target site, thus confirming the sequence specificity of RNAi (Figure 2C, grey bars). All pLAI variants, except the wild-type, were cotransfected with the matching shRNA (Figure 2C, white bars). The R3 and R3? escape mutants are potently inhibited by their matching shRNA and the level of inhibition is comparable to the original situation of the wild-type virus with the wild-type shRNA. For the R4 escape mutant, inhibition is improved with the matching shRNA (white bar) compared to the wild-type shRNA (grey bar), although inhibition is only partially restored. |
| R6 is a special escape mutant because the single nucleotide substitution imposes an altered RNA folding that occludes the target site (Westerhout et al, 2005). Therefore, RNAi is not rescued with the matching shRNA. For the R9 mutant, the matching shRNA is not effective at all. These results show that it is possible to reconstitute at least some level of inhibition, and in most cases restore complete inhibition, with a second generation of shRNAs aimed at escape variants. |
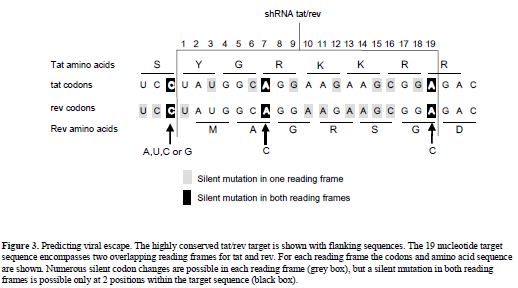
| We can apply this concept to the effective and conserved targets of HIV-1 listed in Table 1. One can empirically determine which target site mutations result in replication competent escape variants by studying virus escape in cell culture infections. For the targets with a conserved sequence, we assume that most point mutations will affect virus replication. A conserved sequence may have multiple functions, e.g., multiple overlapping reading frames and RNA or DNA signals that code for splicing, packaging or promoter functions. Because it is likely that multiple mutations in the target sequence will result in impairment of its function(s), a first assumption is that only a single point mutation is allowed. A second assumption is that only silent codon changes can occur. For example, the conserved tat/rev sequence (Surabhi and Gaynor, 2002) encodes two overlapping reading frames (Figure 3). There are 9 silent codon changes possible for each individual open reading frame, but only at position 7 and 19 these changes are silent in both reading frames (Figure 3). In both cases, a silent variation at the first nucleotide position of an Arginine (R) codon in tat coincides with a silent change of a third nucleotide of a rev codon. A combination of three shRNAs targeting the wt, A7C and A19C sequences may prevent viral escape. |
| In general, this strategy may be applied to all conserved HIV-1 targets. It is important to note that the success of this approach depends upon several criteria. First, only a limited number of escape routes should be possible, otherwise too many second generation shRNAs have to be designed. Second, the new shRNA should be capable of inducing RNAi. For instance, shNEF-wt partially inhibits the escape mutant LAI-R9 but the adapted shNEF-R9 failed to silence LAI-R9 at all, indicating that this particular shRNA is inactive (Figure 2C). Finally, the target site should remain accessible (Westerhout et al, 2005; Schubert et al, 2005b). The R6 escape variant provides an example where any shRNA is likely to be ineffective because this nucleotide substitution induced a change of the RNA structure such that the target sequence is inaccessible (Westerhout et al, 2005). |
| Anti-AIDS gene therapy |
| For an effective therapy, the RNAi-triggering genes have to be transferred into the appropriate cells. Such a gene therapy protocol seems ideally suited for the treatment of individuals that are chronically infected with HIV-1 and fail on standard antiretroviral therapy. In chronically infected individuals, HIV-1 infects a significant fraction of the mature T cells each day, leading to cell killing directly by HIV-1 or indirectly by the HIV-induced immune system. In addition, HIV-1 infection may induce immune activation resulting in apoptosis in uninfected bystander cells (Herbein et al, 1998). Nevertheless, the preferential survival of even a minority of shRNA-expressing cells will result in their outgrowth over time. One could carry out an ex vivo gene therapy treatment of either the patients mature CD4+ T cells from the blood or CD34+ haematopoietic stem cells from the bone marrow, and give them back to the patient. The stem cells will proliferate into mature T cells and move to the periphery, thus forming a constant supply of cells that resist HIV-1 infection. This means that even a relatively inefficient ex vivo gene therapy protocol may be beneficial through partial reconstitution of the immune system. |
 |
| Viral vectors based on HIV-1, the lentiviral vectors, are superior over retroviral vectors for transduction of either T cells or haematopoietic stem cells. These vectors are preferable candidates for gene therapy development for treatment of HIV-1 infected patients (Strayer et al, 2005). However, the introduction of shRNAs against highly conserved HIV-1 sequences may pose a problem for vector production, since these shRNAs could cross-react with critical sequences of the vector. Indeed, it has been reported that expression of shRNAs that target the vector can result in a reduction of the titer (Banerjea et al, 2003; Chang et al, 2005). One may expect that when a multiple shRNA expression lentiviral vector is developed, this will become a serious issue, but solutions for this potential problem are already available. With safety in mind, lentiviral packaging plasmids were developed that are optimized for human codon usage (Kotsopoulou et al, 2000; Wagner et al, 2000), and thus quite different from the original viral sequences. We found that the reduction in titer will be resolved when these constructs are used for lentiviral vector production (Ter Brake and Berkhout, in preparation). |
 |
| shRNA expression lentiviral vector is developed, this will become a serious issue, but solutions for this potential problem are already available. With safety in mind, lentiviral packaging plasmids were developed that are optimized for human codon usage (Kotsopoulou et al, 2000; Wagner et al, 2000), and thus quite different from the original viral sequences. We found that the reduction in titer will be resolved when these constructs are used for lentiviral vector production (Ter Brake and Berkhout, in preparation). |
| Interferon response |
| The paradigm was that only dsRNAs larger than 30 basepairs induce the interferon response, but it was recently shown this is also true for some siRNAs. This effect is largely dose-dependent and may be specific for synthetic siRNAs (Myers et al, 2003; Kim et al, 2004). In addition, for systemic delivery of siRNAs in vivo, a sequence motif that interacts with a Toll like receptor was implicated in the induction of interferon (Hornung et al, 2005). However, these kind of interferon responses were associated with extracellularly added siRNAs and not intracellular siRNAs when tested in peripheral blood mononuclear cells in vitro (Sioud, 2005). We propose to induce RNAi by intracellular shRNA expression and, therefore, we expect that these undesired responses will not occur. |
 |
| Since siRNA transfections clearly differ from intracellular shRNA expression, results obtained in siRNA experiments may not be relevant for our proposed therapeutic approach. Two shRNAs expressed from a lentiviral vector at a relatively high multiplicity of infection (moi) did not induce the interferon response (Anderson and Akkina, 2005). However, the presence of an AA-dinucleotide near the transcription start site of an shRNA was implicated in interferon induction (Bridge et al, 2003; Pebernard and Iggo, 2004). Interestingly, such a shRNA was a more prominent inducer when expressed from a U6 promoter compared to expression from an H1 promoter (Bridge et al, 2003). These experiments used an moi of 10, and the observed interferon response for these shRNAs was lost when an moi of 1 was used. More importantly, the majority of shRNAs did not induce an interferon response even at the high moi. Few of the published anti-HIV siRNAs and shRNAs (Table 1) contain an AA-dinucleotide near the transcription start site, and none contain the motif that interacts with the Toll like receptor. We propose that the multiplicity of infection should always be kept below 1 in a therapeutic setting. This will keep expression levels low, thereby preventing possible side effects such as translational inhibition or saturation of the miRNA or siRNA machinery because these effects are usually associated with a high concentration of siRNA molecules (Scacheri et al, 2004; Huppi et al, 2005). Anyhow, with at least nine candidate RNAi molecules available for the multi-shRNA approach, one could test for putative side effects and select non-toxic shRNAs for a clinical trial. |
| Lentiviral vector design for multiple shRNA expression |
| An important step towards anti-HIV RNAi therapy is the design of a lentiviral vector that expresses multiple shRNAs. The goal is to achieve equal expression of each individual shRNA from the multiple shRNA vector, resulting in similar levels of inhibition per shRNA as compared to single shRNA constructs. In case the absolute inhibition would decrease for individual shRNAs in the multiple shRNA vector, selective pressure would be lost, and therapy would likely fail. |
| An obvious way to construct a multiple shRNA vector is to clone different expression cassettes with the same promoter in tandem orientation (Gonzalez et al, 2005). In the lentiviral vector context, this may present problems because recombination may result in deletion of one or multiple repeats and the intervening sequence (An and Telesnitsky, 2001; Marzio et al, 2001). A solution to this problem would be to increase the number of viral vector copies per cell, thereby ensuring the presence of at least one intact copy. However, this may be undesirable because the concentration of shRNAs will increase and introduce the risk of undesirable side effects. In addition, there will be an increased chance of insertion-induced oncogenesis and a significant increase in treatment cost. |
| To avoid recombination within the lentiviral vector, one could use different promoters for shRNA expression. Nine different promoters have been used to drive the expression of shRNAs, most common are the RNA polymerase III promoters U6 (Yu et al, 2002) and H1 (Brummelkamp et al, 2002), which have been routinely used in HIV-1 research (Paul et al, 2003; Boden et al, 2003a; Scherer et al, 2004; Das et al, 2004; Boden et al, 2004a; Lee et al, 2005; Chang et al, 2005). Alternatives include the RNA polymerase II promoter for the U1 snRNA gene (Denti et al, 2004), the tRNA Val promoter (Kawasaki and Taira, 2003), the RNA polymerase I promoter (McCown et al, 2003), the polymerase II cytomegalovirus (CMV) promoter (Boden et al, 2003b; Song et al, 2004), the modified human tRNA Met promoter (Boden et al, 2003b), the VA I Adenovirus promoter (Cordelier et al, 2003) and the 7S K promoter (Koper-Emde et al, 2004). A direct comparison of each of these promoters in the lentiviral vector context will provide the information to decide which ones to combine in a multiple-shRNA viral vector. |
| Another interesting option to avoid promoter duplication would be to express multiple siRNAs from one expression unit as a single transcript. Two shRNAs connected with a short linker sequence can be equally effective as individual shRNAs (Anderson et al, 2003). In addition, it may be possible to stack functional siRNAs on top of each other, with or without bulges in between. These transcripts could be designed resembling to mimic the branched microRNA precursors (Boden et al, 2004b). However, one must be aware that such complex RNA structures may result in significant problems within the context of the lentiviral vector RNA genome. Approaches like these are still in the early development phase, but could eventually lead to an effective approach for multiple siRNA expression. |
| When first generation shRNAs are combined with second generation shRNAs in the vector design, a shRNA repeat sequence will be present. However, the second generation shRNA differs in at least one basepair from the first generation and can be modified by introducing G-U wobble basepairs and a different loop sequence (Miyagishi et al, 2004). These modifications can limit the repeat sequence to 19 nucleotides, which contains at least one point mutation. We anticipate that such a minimal and imperfect repeat will not be problematic. |
Conclusions |
| We have reviewed current publications involving RNAi approaches using siRNAs or shRNAs against the HIV-1 genome. Since it is known that the application of individual shRNAs can lead to viral escape, the focus of this review is on the development of a durable gene therapy that prevents viral escape. We worked out two strategies that may be employed. First, like many have suggested previously, multiple shRNAs targeting highly conserved HIV-1 sequences can be combined. This will increase inhibition, and reduce the chances of escape exponentially with each added shRNA. Second, we introduce a new strategy to counteract viral escape. When highly conserved sequences are targeted, escape options are limited. If, in addition to the wild-type shRNA, a selection of second-generation shRNAs would be included that anticipate these routes, escape may be prevented. As a proof of concept, we demonstrate that it is possible to efficiently inhibit escape mutants with such a second generation of shRNAs. |
| Which shRNAs should than be used for an RNAi based gene therapy? The incoming HIV-1 RNA genome represents an efficient target and viral messengers are likely to be subject to RNAi in the nucleus and cytoplasm. Therefore, we argued that target site selection can be based solely on conservedness among the different HIV-1 isolates. From all published target sequences from the HIV-1 genome, nine are conserved in at least half of all isolates. These seem the best candidates for a gene therapy based on multiple RNAi molecules. Studying escape for individual shRNAs in viral infection cultures will reveal whether second-generation shRNAs should be included. We expect that risks of interferon induction or other side effects like off-target inhibition or saturation of the miRNA machinery can be avoided in a gene therapy setting when the dosage of vector is kept low. Nevertheless, individual shRNAs and any combination of shRNAs should be tested for these putative side effects. |
| Recently, Sirna Therapeutics reported interim data for phase I clinical trials for treatment of age-related macular degeneration (Quinlan, 2005). No adverse or toxic effects were observed and disease stabilized in all patients. In addition, the first clinical trial involving lentiviral vectors showed no adverse effects in five patients (Manilla et al, 2005). Combined, these results pave the way for a clinical trial involving RNAi and lentiviral vectors. Indeed, John Rossi of the City of Hope Beckman Research Institute in California in collaboration with the Australian company Benitec Limited are planning a clinical trial involving a multi-RNA lentiviral vector approach in HIV-1 infected patients (Check, 2005). Their vector encodes a TAR decoy, a ribozyme against CCR5 and also a single shRNA against the HIV-1 genome (Li et al, 2003; Akkina et al, 2003; Li et al, 2005). The outline we presented here may form the basis for the development of the next phase in RNAi-based treatments of HIV-1 infection. |
Acknowledgements |
| We thank Thijn Brummelkamp for the pSUPER plasmid and Ellen Westerhout for the pLAI constructs. We thank Stephan Heynen for performing CA-p24 Elisa. RNAi research in the Berkhout lab is sponsored by NWO-CW (TOP grant) and Senter (grant with Viruvation BV). |
Statement of Compteting Interests |
| The authors declared no competing interests. |
References
|