- Journal of RNA and Genomics (2005) Review Article
Mechanisms of RNAi: mRNA cleavage fragments may indicate stalled RISC
| Torgeir Holen* Centre for Molecular Biology and Neuroscience (CMBN), University of Oslo, Norway |
| *Correspondence to: Torgeir Holen, Email: torgeir.holen@basalmed.uio.no, Tel: +47 2285 1018, Fax: +47 22851488 |
| (Received 26 April 2005; Revised 15 May 2005; Accepted 18 May 2005, Available online 28 July 2005; Published 12 August 2005) |
Abstract
The molecular mechanism of RNA interference (RNAi) is under intense investigation. We previously demonstrated the existence of inactive siRNAs and also of mRNA cleavage in vivo in human cells. Here it is shown that some siRNAs with low activity leave mRNA cleavage fragments while an siRNA with higher activity does not. The pattern is consistent with both short-term (4-24 hours) and long-term (1-4 days) time-series. Analysis of the putative 3’ mRNA cleavage product showed high GC content immediately after the cleavage point. The cleavage fragments might indicate a stalled or slowed RNAi cleavage complex - possibly in the RISC enzyme restoration phase - and thus constitute a novel explanation for the existence of inactive siRNAs.
Keywords |
| RNAi, RISC, siRNA, activity, mechanism |
Introduction |
| RNA interference (RNAi) is a cellular process whereby introduced double-stranded RNA can induce degradation of target RNA with sequence similarity (Fire et al, 1998). The double-stranded RNA is broken down into small, ~21- mer RNA (Hamilton and Baulcombe, 1999), by the RNase III-like protein Dicer (Hammond et al, 2000). RNAi has become a very widely used tool for functional genomics after synthetic small interfering RNA (siRNA) was shown to be able to induce RNA degradation also in mammalian cells (Elbashir et al, 2001a). |
| The small RNA is incorporated into the RNA-induced silencing complex (RISC) (Bernstein et al, 2001). RISC has been shown to be a multi-turnover enzyme (Hutvagner and Zamore, 2002; Haley and Zamore, 2004), The enzyme has to be restored between enzymatic cycles (Haley and Zamore, 2004). RISC consists of several components, some of which are still uncharacterized, and genetic screens in lower organisms have unearthed a long list of candidate RNAi genes (Meister and Tuschl, 2004; Tang, 2005). Target degradation seems to be initiated by mRNA cleavage, both in Drosophila and in mammalian cells (Elbashir et al, 2001b; Holen et al, 2002). A central candidate for being the mRNA endonuclease “Slicer” is the Argonaute protein Ago-2 (Meister et al, 2004; Liu et al, 2004; Rivas et al, 2005). |
| Some siRNAs are inactive with the activity having strong sequence dependence, since a small shift in position of only 6 nucleotides can give rise to an inactive siRNA from a highly active siRNA (Holen et al, 2002). Statistics on large siRNA screening studies showed that active siRNAs had a bias towards a low GC content in the 5’ end of the antisense strand (Khvorova et al, 2003). One mechanistic explanation for the existence of inactive siRNAs is that this asymmetry in the ends of the duplex leads to differential loading into RISC of one RNA strand over the other, in Drosophila embryo lysates (Schwarz et al, 2003). |
| The results presented here suggest that another mechanism for inactive siRNAs could be the slow restoration of RISC complexes – and thus the accumulation of mRNA cleavage products – perhaps due to the high GC content in the 5’ end of the siRNA. |
Materials and Methods |
| Preparation of siRNA |
| The 21-mer single stranded RNAs were synthesized at 20 nmol scale by Ambion (Austin, Texas, USA). The RNA strands were resuspended in nuclease-free water (Ambion) and the concentration of the strands was verified by spectrometry using NanoDrop (Saveen Werner). Annealing of the complementary strands was performed by mixing equal amounts of RNA and briefly heating the mixture to 65oC for 5 min and then allowed to cool to room temperature over 5 min. Annealing was verified by running siRNA on non-denaturing 12% (w/v) PAGE gels, staining with SYBRgold (Invitrogen) and visualizing bands by fluorescent scanning on a Typhoon 9410 (Amersham Biosciences). |
| Cell culture and transfection |
| HeLa cells were maintained in Dulbecco’s minimal essential medium (DMEM) supplemented with 10% (v/v) foetal calf serum (Gibco BRL). The cells were passaged at subconfluence and plated 24 hr before transfection in 6-wellplates, where each well is approximately 10 square centimeters. Transfections were performed using 1.44 Lg siRNA per well and 3.6 Ll Lipofectamine2000 (Invitrogen), and thus a ratio of 2.5 of Lipofectamine2000 (in microliters to micrograms of RNA). |
| Isolation of mRNA and northern blots |
| The mRNA was isolated at various times after transfection using Dynabeads oligo(dT)25 (Dynal) and then fractionated at 1.5 % (w/v) agarose gels containing 0.8 M formaldehyde for 3 hr at 50 V. The agarose gel was then washed for 2 x 15 min in distilled water before the RNA was blotted onto pre-wetted BrightStar-Plus nylon membranes (Ambion). The Fen1 mRNA was detected using a radioactive probe corresponding to the protein coding region (373- 1515, relative to Fen1 mRNA accession number NM_004111), using random priming with Rediprime II (Amersham Biosciences) and P-32 alpha-CTP (Amersham Biosciences). Hybridization was performed overnight using ExpressHyb (BD Biosciences), with four subsequent washes at room temperature and 50 degrees with Wash Buffer I (2 x SSC + 0.05% , w/v, SDS), and with Wash Buffer II (0.2 x SSC + 0.1%, w/v, SDS), respectively. |
| The radioactive signals were visualized with a Phosphor storage Screen and a Typhoon 9410 imager (Amersham Biosciences). Quantification of relative signal strength performed with the ImageQuant software (Nonlinear Dynamics, Amersham Biosciences). |
Results and Discussion |
| We previously investigated inactive siRNAs using relatively sensitive and quantitative Northern blots (Holen et al, 2002; Amarzguioui et al, 2003; Holen et al, 2003). While screening for siRNAs against the DNA repair gene Flap-endonuclease 1 (Fen1) in a new study (Figure 1A), faint, 3’ cleavage products were observed with two of the siRNA species when harvesting the mRNA at 24 hr posttransfection (data not shown). |
| The presence of these mRNA cleavage fragments was an interesting observation and, therefore, this was investigated further. When improving the quality of northern blots and accentuating the signal difference between highly active and less active siRNAs by isolating mRNA at 12 hr post-transfection, the cleavage products were clearly observed only in the lanes of two less active siRNAs, Fe668i and Fe884i, while our best candidate, Fe775i, had no such cleavage product (Figure 1B). Our previous hypothesis regarding such cleavage products proposed that cleavage products were due to lack of mRNA-degradation capacity in a down-stream ratelimiting step (Holen et al, 2002). The new data contradict this hypothesis. |
| The electrophoretic migration of the cleavage fragments was measured. Using the main Fen1 band and the loading control GAPDH, with mRNA sizes at 2.3 kb and 1.3 kb respectively, the sizes of the cleavage products were calculated to be consistent with primary cleavage at the siRNAsites (data not shown). |
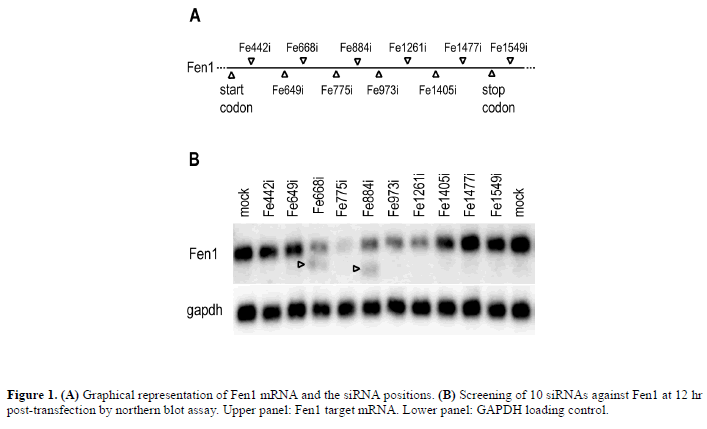 |
| A time-series study was performed to investigate whether the cleavage fragments were of a transient nature. It seemed possible that perhaps Fe775i had produced and already lost such a cleavage fragment while depleting the Fen1 mRNA pool five-fold sometime during the first 12 hr post-transfection. The mRNA was harvested at time points 4 hr, 8 hr, 12 hr and 24 hr for Fe775i and Fe668i (Figure 2). At no time was a cleavage product observed in the Fe775i-samples, while with Fe668i a gradual build-up of cleavage product was observed (Figure 2). |
| While studying the long-term silencing over several days, it was found that the main Fen1 mRNA band recovered to full strength for the weaker siRNA candidate, Fe668i, while our best siRNA candidate, Fe775i, retained more than 60% silencing even after 4 days (Figure 3). At 48 hr the cleavage product was in fact much stronger than the main band of the Fe775i-sample (Figure 3). |
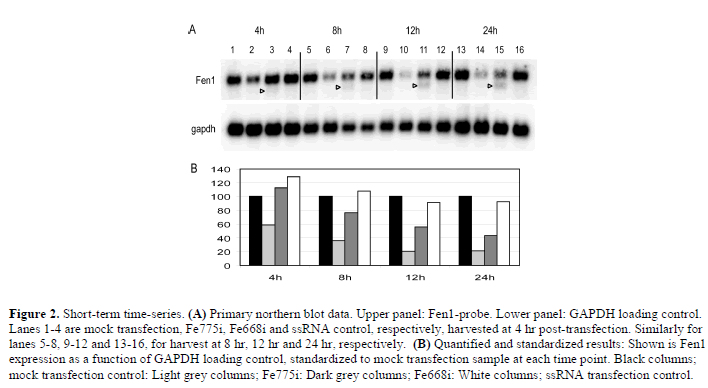 |
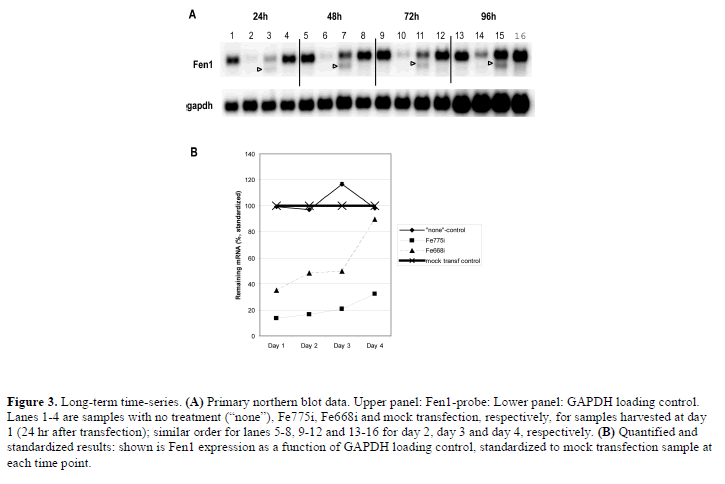 |
| We also observed in a previous study that the mRNA depletion by RNAi seems to be finely balanced with mRNA production (Holen et al, 2002). The cleavage product continues to be produced even after the net mRNA silencing has been overwhelmed by mRNA transcription. As the last Fe668i-silencing is lost between 72 hr and 96 hr, cleavage product continues to build up (Figure 3). This demonstrates that putatively inactive siRNAs can also have some activity, although too low compared to the mRNA transcription rate to affect the steady-state concentration of mRNA. If the cleavage product is associated with stalled RISC – as would seem to be necessary to protect a cleaved mRNA from general degradation – this would be a novel mechanism resulting in less active siRNA species, such as Fe668i and Fe884i. |
| Using Martinez and Tuschl’s proposed cleavage point (after the 10th nucleotide counting from the 5’ end of the antisense strand) (Martinez and Tuschl, 2004), the GCcontent was tabulated and analyzed in the putative 3’ mRNA-cleavage products, finding that Fe668i and Fe884i (but not Fe775i, nor Fe973i and Fe1261i, two other weak siRNAs) had a high GC content in the first 8 base-pairs after the cleavage point (Figure 4). |
 |
| Efforts to find stable hairpin structures – an alternative hypothesis for the existence of cleavage products – in the cleaved mRNA were fruitless. One might postulate such a stabilizing hairpin structure somewhere downstream of the cleavage points of Fe668i and Fe884i, but if so, it would be reasonable to expect the upstream siRNA of Fe775i to also produce the same cleavage products. However, Fe775i does not produce such cleavage products. Furthermore, some results from our previous study can now be reinterpreted in the light of these new observations. Two central GC-pairs were mutated, with a double and single mutation, hTF167-M1 and hTF167-M2 (Holen et al, 2002). These mutations reduced the mRNA activity but the single and double mutations still retained approximately 80 and 70 percent mRNA depletion, respectively. Both the single and the double mutation were situated 5’ of the putative cleavage point. Interestingly, the cleavage product was partly lost with M1, and completely lost with M2. |
| At the time we interpreted the loss of cleavage product as a consequence of the lower silencing capacity and in the light of the hypothesis of a saturated downstream ratelimiting step in the RNAi mRNA depletion process. However now this loss of cleavage products can be reinterpreted to support the hypothesis of RISC restoration being dependent on the binding strength of the antisense siRNAmRNA duplex after cleavage. |
Conclusions |
| In conclusion, these observations suggest that the strandselection hypothesis (Schwarz et al, 2003) may not be the sole explanation for the inactivity of siRNAs. The cleavage fragments observed with a series of siRNAs indicate that the restoration of RISC complexes after cleavage of mRNA can also be a rate-limiting process, which seems to be affected by the siRNA sequence, possibly the GCcontent of the 5’ end of the antisense siRNA strand. |
Acknowledgments |
| This work was supported by Arne Klungland and Ole Petter Ottersen Laboratories. |
Statements of Competing Interests |
| The author declared no competing interests. |
References
|