Research Article - Journal of Genetics and Molecular Biology (2022) Volume 6, Issue 4
Unconventional droplet digital PCR assays for SMN1/SMN2 assignment of pathogenic point mutations.
Michela Bulfoni1*, Elena Betto2, Lorenzo Verriello3, Maria Elena Pessa3, Daniela Cesselli1,4, Francesco Curcio1,2 and Incoronata Lonigro1,2
1Department of Medicine, University of Udine, Udine, Italy
2Department of Biomedical Science and Technology, Institute of Clinical Pathology, ASUFC, Udine, Italy
3Department of Neurosciences, Neurology Unit, ASUFC, Udine, Italy
4Department of Medical and Biological Sciences, Institute of Pathology, ASUFC, Udine, Italy
- *Corresponding Author:
- Michela Bulfoni
Department of Medicine
University of Udine, Udine, Italy
E-mail: michela.bulfoni@uniud.it
Received: 08-Jun-2022, Manuscript No. AAGMB-22-66127; Editor assigned: 09-Jun-2022, PreQC No. AAGMB-22-66127(PQ); Reviewed: 06-July-2022, QC No. AAGMB-22-66127; Published: 20-July-2022, DOI:10.35841/aagmb-6.4.116
Citation: Bulfoni M, Betto E, Verriello L, et al. Unconventional droplet digital PCR assays for SMN1/SMN2 assignment of pathogenic point mutations. J Genet Mol Biol. 2022;6(4):116
Abstract
Almost 4% of spinal muscular atrophy affected patients (SMA) carry the deletion in one copy of the Survival Motor Neuron gene 1 (SMN1) and a deleterious point mutation on the other copy of the gene. Correct assignment of the point mutations to the disease causative gene is not a rapid procedure due to the existence of the twin Survival Motor Neuron gene 2 (SMN2) not involved in the onset of the disease. For many years, transcripts sequence, cloning and clone screening were the standard procedures for establishing the unequivocal localization of a point mutation within SMN1 or SMN2 allowing proper diagnosis for symptomatic subjects and carrier testing for relatives at reproductive risk. Many clinical diagnostic laboratories have recently adopted long-range PCR and Sanger sequencing for the identification of the deleterious point mutations, splicing variants and small indels in specifically SMN1. However, the techniques mentioned above are somewhat time-consuming, while great benefits can derive for children with SMA from an early diagnosis and an early entry into a therapeutic trial. The method we developed relay on ddPCR technology and gene- and SNP-specific TaqMan MGB probes to simplify experimental procedures and reduce the turnaround time to reach the correct diagnosis. In addition, this method has the potential to have a more general application in detecting mono or bi-allelic segregation of deleterious coding point mutations in many other recessive inherited diseases, such as some of the results obtained through NGS based tests, without the need to analyze parents or other relatives of the patient.
Keywords
Spinal Muscular Atrophy (SMA), Droplet digital PCR (ddPCR), SNP co-segregation, Gene discrimination, Genetic diagnosis, Biotechnology, Multiplexing, High-throughput molecular diagnostics.
Introduction
Spinal muscular atrophy (SMA) is the second most frequent lethal autosomal recessive disease in Europeans next to cystic fibrosis with an incidence of 1/6000 - 1/10,000 and an average carrier frequency of 1/40 healthy subjects[1–3]. The disease is characterized by progressive proximal muscle weakness and atrophy due to progressive death of alpha-motorneurons in the anterior horn of spinal cord and affected subjects may exhibit varying degrees of disease severity (SMA Type 0- SMA type IV). The primary biological cause of the disease lies in the insufficient synthesis of functional survival motor neuron protein (SMN protein) and the complete absence of the protein does not allow the survival of the embryo, which undergoes spontaneous abortion in the first trimester of pregnancy [4]. The SMN protein is a component of large macromolecular complexes that are found both in cytoplasmic and nuclear compartments and plays a crucial role in the assembly of RNP subunits of the spliceosome and thus in mRNA biogenesis [1]. The SMN protein is encoded by the SMN gene present in 5q13 in two highly homologous copies, a telomeric gene (SMN1) and a centromeric one (SMN2) that differ in five basepairs, two of which localized in exonic regions (exon 7 and 8). Only one of the two exonic changes (exon 7) is causative of the huge functional difference between the SMN1 and SMN2 genes. Precisely, the critical change consists in a C to T substitution at +6 position of exon 7 that does not allow the efficient inclusion of this exon into the SMN2- derived transcripts. In a healthy subject almost 90% of the SMN full-length transcript that includes exon 7 and results in a functional protein derives from SMN1 while SMN2 provides the remaining 10% [1,5].
Therefore, SMN1 is the disease causative gene and SMN2 is considered a phenotypic modulator always present with variable copy number in the affected subjects [1,6]. SMA individuals present a homozygous deletion in SMN1 or conversion of SMN1 in SMN2 in approximately 96% of cases. The remaining 4% of SMA patients are compound heterozygous of the deletion or conversion on one allele and an intragenic mutation on the second one. Actually, the identification of SMN1 deletion or gene conversion is well supported especially through relative quantification techniques such as simplex or multiplex Real-Time PCR [7,8]. Multiplex Ligation-dependent Probe Amplification (MLPA) [8–11]. On the contrary, the identification of deleterious point mutations in the SMN1 coding sequence of a compound heterozygous patient or a healthy carrier requires the SMN mRNAs cloning and colony screening or alternatively requires gene specific long-range PCR (LR-PCR) followed by Sanger sequencing to recognize which transcript, SMN1 or SMN2, carries the mutation[12–15]. Procedures such as transcript cloning are quite tedious for the operator, time consuming and therefore not desirable for prenatal or neonatal diagnosis as well as for carrier screening of relatives at reproductive risk. LRPCR is a powerful technology aimed to identify new SMN1 gene mutations also deep in introns [14]. However, the great majority of SMN1 mutations identified up to now are in exons or near the exon/intron boundary, as reported for example in the study conducted by Alias et al. 2009 and computational structural analysis illustrating how do coding mutations of SMN1 lead to structural/functional deficiency of the SMA protein [15,16].
SMN1 coding point mutations or micro-insertions/deletions can be easily identified by standard Sanger sequencing using genomic DNA and can be easily identified by standard Sanger sequencing using genomic DNA. What remains still time consuming is the assignment of the identified deleterious point mutation to SMN1 or SMN2. The presence of the point mutation in a single copy of SMN1 per diploid genome of an affected subject, will confirm the clinical diagnosis and targets the mutation for other relatives at reproductive risk within the family. Moreover, and above all, the speed in reaching the definitive diagnosis is fundamental for the therapeutic intervention in children with SMA made possible today. Given the therapeutic importance of identifying quickly and with certainty the co-segregation of a deleterious SNP with SMN1 or with SMN2, we have developed an analytical technique that we think may be useful for this purpose.
Here, we describe a method based on digital droplet PCR technology (ddPCR) for a rapid co-segregation study of natural SNPs in exon 7 of SMN1 and SMN2 and pointmutations identified elsewhere in the coding region of these genes. The ddPCR is a third generation PCR method with the powerful ability to determine the absolute concentration of nucleic acids (DNA/RNA) and identify rare allelic variants, non-abundant RNA transcripts and copy number variations (CNVs) without the need of an external standard curve [2– 4,6,17–20]. In ddPCR, the target template is partitioned in up to 20,000 micro droplets, each containing either a single copy or no target nucleic acid. The amplification of a target molecule within a single droplet, as an individual PCR microreactor, allow to achieve the defined quantity of a specific initial nucleic acid and help in locate a specific variant on the target template.
The amplification of the target sequence is independently carried out in a single droplet in the presence of a targetspecific probe conjugated with a fluorophore. In this way, droplets containing amplified target sequences are detected by fluorescence and are discriminated from those where amplification has not occurred. In each droplet, the absolute number of target nucleic acid molecules can be measured using binomial Poisson statistics and defined as template concentration directly from the ratio of positive to total droplets [4,5,18,21–23]. However, in some cases, a single compartment could contain more than one target molecule, and in this case the count should be corrected with Poisson statistics.
Furthermore, the ability of the new platforms for multiplexing, has enhanced ddPCR standard applications [7,24–26]. In fact, in multiplexing assays different probe concentrations can be mixed to obtain a multi-target analysis per single sample using only 2 filters. The possibility to discriminate more targets in a single dot-plot graph is based on the differential position of droplets caused by different fluorescent label concentration of the probes [7,24,26]. Due to its properties of high sensitivity (1/10,000), reproducibility, and accurate allele classification, ddPCR technology has proven to be the diagnostic tool of choice [18–20,22,23] especially in pathological conditions with somatic mosaicism such as cancer, allowing the identification of very low mutant allele frequencies. Recently, ddPCR has been commonly applied for efficient characterization of circulating tumor DNA (ctDNA) profile, to monitor treatment response, and relapse in real time [19,26-31]
Different studies, successfully show that ddPCR can be used for SMA diagnostic intents in order to measure SMN1 copy number using RPP30 gene as reference, starting from genomic DNA [32]. Validation studied have demonstrated the validity of the method with respect to MLPA [33]. The lack of a standard curve for the absolute quantification of SMN1 or SMN2 copy number and the high sensitivity and specificity for the identification of SMA carrier status, are some of the highlighted benefits of ddPCR. Moreover, ddPCR is useful to predict phenotypic severity of SMA patients by determining the copy number of SMN2 in clinical laboratories. [32-34]
In this work we describe an unconventional analytical method based on the combination of ddPCR technology with TaqMan MGB fluorescence probe chemistry to localize a deleterious point mutation in SMN1 or SMN2 coding region in exon 3, causing protein truncation, identified in an affected SMA patient and an unrelated healthy carrier. We were also able to quantify the amount of the disease-causing mutant transcript, drawing the conclusion that the identified point mutation was also the cause of the low SMN1 derived transcript level. The molecular assay strategy we developed significantly improves assay performance for rapid assignment of coding point mutations in SMN1 or SMN2 genes using only two fluorophores.
Materials and Method
Subject enrolled
Three subjects were enrolled in this study: A SMA type 3A female, an unrelated SMA carrier male and an unrelated negative healthy control. All subjects were treated according to the rules of the Regional Ethics Committee and written informed consent for genetic analysis needed for the research was collected.
SMN genes copy number quantification and sequencing
The three subjects enrolled in this study were genotyped at SMA locus. The genomic DNA has been isolated from peripheral blood using the Qiagen Gentra Puregene Blood Kit according to the manufacturer’s instructions.
The SMN1/SMN2 copy number quantification was performed by multiplex Real-Time PCR [2] using a LightCycler® 480 Instrument (Roche) and MLPA assays (P021 Kit, MRCHolland) according to the manufacturer’s instructions. Capillary electrophoresis of the MLPA products was performed using an ABI 3500 sequencer (Applied Biosystems) and results were analyzed using Coffalyser software (MRCHolland). Both methods yielded consistent results. For sequence analysis of SMN1 and SMN2 of the SMA subject and healthy carrier, the nine exons and exon/intron junctions of both genes were amplified and sequenced using standard procedures. Briefly, PCR amplicons of SMN genes were obtained using forward and reverse oligonucleotides (Sigma Aldrich) 5' tailed with M13 sequence to simplify the next sequence step. The sequence reaction was performed following standard procedures on an ABI 3500 Genetic Analyzer (Applied Biosystems) automated sequencer.
PBMC isolation and RNA extraction
For RNA extraction, peripheral blood mononuclear cells (PBMCs), collected from the SMA patient, SMA healthy carrier and healthy control were isolated from EDTA blood samples by density gradient centrifugation, using the Ficoll®Paque Plus (Sigma Aldrich). PBMCs were washed twice with PBS; cellular pellets have been collected and subjected immediately to RNA extraction or frozen at -80°C.
Total RNA was extracted from PBMCs using the RNeasy Mini Kit (Qiagen), following the manufacturer's instructions. Briefly, cells were lysed and homogenized using the RTL buffer and then RNA purification was carried out on silicabased columns with microspin technology. RNA was finally eluted in 50uL of nuclease-free water. The Nanodrop 2000 (Thermo Fisher Scientific) spectrophotometer was used to quantify the amount of total RNA recovered.
DNAse digestion and cDNA synthesis
To remove traces of genomic DNA, the isolated RNA was treated with DNAse I (Qiagen, RNase-Free DNase Set), accordingly to the manufacturer's instructions. Then, the mRNA was reverse-transcribed into cDNA using oligoDT primers and the SuperScript III reverse-transcriptase enzyme (all from Invitrogen, Life Technologies). The choice to reverse transcribe using oligoDT primers was made to obtain a fulllength transcript molecule in each droplet. The incubation profile was as follow: 25°C for 10 min, 42°C for 50 min and 85°C for 5 min.
Primer and Probe design
Primer and TaqMan MGB probe combinations for SNP analysis were designed for the loci of interest using as templates the SMN1 mRNA NM_000344.4 and the SMN2 mRNA NM_017411.4 by the prime PCR tool (Biorad) and synthesized by ABI/Thermo-Fisher Scientific company. Primers annealed to cDNA at a temperature of 60°C, according to the manufacturer's recommendations (Thermo- Fisher Scientific). Sequences of all probes and primers for ddPCR assays are reported in Table 1.
| Primer/Probe | Sequence |
|---|---|
| SMN1/2 ex 7 FW | 5’-CATGAGTGGCTATCATACTG-3’ |
| SMN1/2 ex 7 RV | 5’-ATTTAAGGAATGTGAGCACC-3’ |
| SMN1 ex 7 probe | 5’-[VIC]-TTTTGATTTTGTCTgAAACCC-[MGB]-3’ |
| SMN2 ex 7 probe | 5’-[VIC]-TTTTGATTTTGTCTaAAACCCAT-[MGB]-3’ |
| SMN1/2 ex 3 FW | 5’-TTTCCCCAATCTGTGAAGTA-3’ |
| SMN1/2 ex 3 RV | 5’-ATCTGTTGAAACTTGGCTTT-3’ |
| SMN ex 3 WT probe | 5’-[FAM]-ATTTTCATTCTCTTgAGCAT-[MGB]-3’ |
| SMN ex 3 MUT probe | 5’-[FAM]-ATTTTCATTCTCTTaAGCAT-[MGB]-3’ |
Table 1. Primer and probe sequences used in the ddPCR assay for the detection of point mutations on SMN1 and SMN2 transcripts.
Experimental set-up
For experimental set-up, optimisation of assay parameters such as TaqMan MGB probe concentration, annealing temperature, and amplification efficiency conditions was performed. To differentiate the amplitude between negative and positive droplets and to reduce the background signal, we performed a temperature gradient in the annealing step, testing a temperature range from 55° to 60°C (Data not shown).
The fluorescence emission of TaqMan MGB probes was tested individually, running different single reactions. Specifically, we performed a triplicate assay for both, exon 7 of SMN1 and exon 7 of SMN2 using a gene-specific VIC-conjugated probe, and for wild-type exon 3 and mutant exon 3 variants using a variant-specific FAM-conjugated probe.
Droplet digital PCR assay (ddPCR)
The ddPCR reaction mixture included 50ng of cDNA template, 2X ddPCR Supermix for Probes (No Dutp; BioRad), 1μL of mixture of primers and probes against the specific targets (respectively 900nM and 250nM), in a final volume of 20uL. As convention, 3 technical replicates per patients were done and opportune internal negative controls were prepared in order to exclude contaminations. Droplet generation was performed using the QX200 Droplet generator (BioRad). The thermal cycling conditions were: 95 for 10 min; 40 cycles of 94 for 30 sec. and 60 for 1 min; 98°C for 10 minutes; 4°C hold, with a ramp rate of 2°C/second and a heated lid at 105°C. Results were analyzed on a QX200 reader (BioRad) using the Quanta Soft software (BioRad).
Data Analysis
Positive droplets, containing amplification products, were discriminated from negative ones by applying a fluorescence amplitude threshold. The reactions with more than 10,000 droplets per well were used for the analysis. The copy number concentration of each sample was reported automatically by the ddPCR software after applying the positivity threshold (Tables 2 and 3). The Poisson error and total error were calculated by QuantaSoft software.
| SUBJECT | SMN1/2 COPY NUMBER | SMN1 total mRNA copies (green+orange droplets) |
SMN total mRNA copies (green+orange droplets) |
%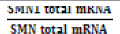 |
| HEALTHY CONTROL | 2:1 | 919 | 1022 | 90% |
| HEALTHY CARRIER | 2:1 | 1175 | 1529 | 77% |
| PATIENT | 1:3 | 190 | 4382 | 6,3% |
Table 2. Proportions of SMN1 and SMN1 mutant transcripts were estimated by calculating the % ratio.
| SUBJECT | SMN1/2 COPY NUMBER | SMN1 EX3-mutant copies (blue+orange droplets) |
SMN EX7-total copies (green+orange copies) |
% |
|---|---|---|---|---|
| HEALTHY CONTROL | 2:1 | 0 | 2093 | 0% |
| HEALTHY CARRIER | 2:1 | 65 | 1179 | 5,5% |
| PATIENT | 1:3 | 184 | 4280 | 4,3% |
Table 3. Proportions of SMN1/2 and SMN1 EX3 & EX7 copies were estimated by calculating the % ratio.
Results
Clinical features and genotyping of subjects at SMA locus
The SMA patient is a female who was 34 years old at the time of molecular testing. She has been in a wheelchair since the age of 10 with a clinical diagnosis of SMA type 3A. Until the age of 14 she was still able to stand up with a minimal aid. So far, no respiratory problems or significant joint retractions have been evidenced, voice is normophonic. Patient manifests: dorsal scoliosis, lower right limb shorter than the left, lingual fasciculations, postural tremor in the hands, paresthesia, marked hypotone and muscle hypotrophy. Ligamentous laxity, mostly distal. Semi quantitative genetic testing was performed as described in the methods section and revealed a SMN1/SMN2 copy numbers of 1/3. Sequence analysis of the exons end exon/intron junctions of the SMN genes was performed and a point mutation was identified in exon 3 of SMN genes. This mutation (c.469C?T; CM081442 Seq. Ref. NM_000344) has been reported before [3] and is a known pathogenic variant that results in the replacement of the codon for a Glutamine residue at position 153 of the protein with a stop codon (p.Gln157X), thus causing a premature interruption of the protein.
The same mutation was identified in the SMN genes of a healthy 20-year-old male. This subject belongs to another family and underwent genetic testing to evaluate his reproductive risk because a first cousin with SMA was identified as a compound heterozygote of deletion/point mutation in exon 3 of the SMN gene/s. The SMN1/SMN2 genotype ratio of the healthy carrier of point mutation is of 2/1.
The healthy subject, used as negative control for SMA, is a 60-year-old unrelated man selected among blood donors of the University Hospital of Udine (ASUFC). This subject has a negative family history for SMA and was found to have a SMN1/SMN2 genotype of 2/1. No sequencing of his SMN genes was performed.
Testing TaqMan MGB probes for SMN1/SMN2 transcripts detection and wild-type and mutant transcripts discrimination in a ddPCR-dependent assay
The aim of this work is to address the use of ddPCR technology and TaqMan MGB probe chemistry for accurate and rapid identification of deleterious SNPs to SMN1 or SMN2 responsible for the onset and severity of the SMA disease phenotype. For this purpose, a specific ddPCR assay was developed for the detection and correct SMN1/ SMN2 assignment of the deleterious point mutation in exon 3 previously identified in the SMA affected subject and the healthy carrier recruited in this study. Since the new assay will be successful only if efficient discrimination of SMN1 and SMN2 as well as of wild-type and mutant alleles occurs, we decided to adopt the TaqMan MGB probes given their known high sensitivity to SNPs. During the experimental set-up, the fluorescence emission of TaqMan MGB probes (VIC or FAM conjugated probes) was tested individually, running different single reactions (see the “Methods” section).
These steps were critical to improve the sensitivity and reproducibility of the designed protocol considering the low expression of SMN1/SMN2 target genes and considering that exon 7- or exon 3-specific TaqMan MGB probes differ by only a single nucleotide. Finally, the optimal probe concentration for both VIC and FAM signals, and the amplification conditions were defined as reported in the “Materials and Methods” section.
The overall results obtained from single probe reaction are summarized in Figure 1, where one dimensional (1D) plots are shown for the VIC channel (A, B panels) and the FAM channel (C, D, E, F panels) individually. Results obtained with healthy control cDNA and separate use of exon 7 SMN1- specific or exon 7 SMN2-specific VIC-labelled probes are shown in panels A and B. The negative droplet cluster (black dots) for the VIC channel was about 1200 fU. Interestingly, two different positive droplet clusters emerged for each individual SMN-specific probe due to cross hybridisation for each TaqMan MGB probe with both target genes (SMN1 and SMN2). This condition was determined by the chemistry of these probes. In the presence of a mismatch, the probe cleavage time during PCR amplification is reduced resulting in a loss of emitted fluorescence intensity [4]. A subsequent generation of two clusters of VIC positive droplets (green points), with different fU intensity is detected. Specifically, a smear with higher fluorescence is generated representative of: i. target-specific binding of the SMN1 probe on the SMN1 transcript in the red box of panel A; ii. target-specific binding of the SMN2 probe on the SMN2 transcript in the blue box of panel B. In contrast, a group of non-specific target droplets, with a lower VIC signal intensity, due to cross-hybridization of the probes with the non-specific target, is generated and is displayed in the lower area of A and B panels. That is, the fluorescent droplets in the blue box of panel A are due to the SMN1 probe on SMN2 transcripts while the fluorescent droplets in the red box of panel B are due to the SMN2 probe on SMN1 transcripts, respectively. These results demonstrate that we can identify the 2 different SMN transcripts and determine their relative abundance using a single pair of primers and a unique TaqMan MGB probe. In a real-time PCR assay, the minimal overlap between target-specific and targetnonspecific signals is automatically removed by the analytical software. In ddPCR assays, all signals derived from single droplets are detected and read, even if they are generated by non-specific binding. This aspect represents an advantage for the operator to study gene-specific co-segregation of SNPs, as we demonstrate in the next section.

Figure 1: Specificity of ddPCR probes for SMN1 and SMN2 transcripts and exon 3 wild-type and mutant transcripts.
The control sample, divided by a vertical yellow line, was tested using both SMN1 (panel A) and SMN2 (panel B) specific probes. Panel A red square = VIC-fluorescence of the SMN1 probe on the SMN1-specific target; Panel A blue square = VIC-fluorescence of the SMN1 probe on the SMN2- nonspecific target; Panel B blue square = VIC-fluorescence of the SMN2 probe on the SMN2-specific transcript; Panel B red square = VIC fluorescence of the SMN2 probe on the SMN1-nonspecific target. Control sample (panels C, D) and healthy carrier sample (panels E, F) were used to test wildtype and mutant exon 3-specific FAM-labelled probes. Panels C and E = FAM-fluorescence of wild-type exon 3 probe on SMN1/SMN2 transcripts (blue dots); Panels D and F = FAM-fluorescence of exon 3 mutant probe on SMN1/SMN2 transcripts (blue dots). No positive events emerged using the exon 3 mutant probe on the control subject transcripts (panel D) whereas positive events were detected in the healthy carrier cDNA analyses (panel F). Black dots represent empty droplets generated in each ddPCR reaction.
Similarly, two independent ddPCR reactions were carried out for discrimination of exon 3 wild type and mutant transcripts and the results are shown in panels C-F of Figure 1. The background signal generated by TaqMan MGB FAM-labelled probes was about 3500-4000 fU. This fluorescence threshold was able to differentiate between empty (black dots) and amplified (blue dots) droplets. FAM-positive signals, relative to exon 3-wild-type and mutant SMN transcripts, showed variable fluorescence intensities, between 5000 and 12000 fU, depending on the gene-specific or variant-specific transcript abundance of each subject analyzed. Representative results obtained from two independent ddPCR reactions conducted on the healthy carrier subject using the exon 3 wild-type specific (panels C, E) and exon 3 mutant-specific (panels D, F) FAM-labelled probes, respectively, are shown in Figure 1. As expected, only wild-type exon 3 signals, presented as blue droplets in figure 1D, were found in the SMN1/SMN2 transcripts of the healthy control subject (panels C, D) while wild-type and mutant exon 3 positive signals were both found in the SMN1/SMN2 transcripts of the healthy carrier subject (panels E, F).
By combining the TaqMan MGB probes for SMN1/SMN2 and wild-type exon 3 transcripts and mutant transcripts together in a multiplex assay, the ddPCR output can be displayed as a 2D-graph. Figure 2 shows a representative 2D-plot relative to the multiplex ddPCR assay performed with the probe specific for SMN1 exon 7 and the probe specific for wild-type SMN exon 3, using the healthy control cDNA as template. The typical result produced is a scatter plot in which the specific targets (SMN1 or SMN2 exon 7 in the VIC channel and exon 3 in the FAM channel) can be discriminated and quantified based on position in the quadrant taken by the positive droplets. SMN exon 7 is plotted on the x-axis while wild-type exon 3 is plotted on the y-axis. In Figure 2, a partition can fall into one of four possible clusters: negative partitions containing no amplified targets (black droplets), partitions containing only positive transcripts for SMN1/SMN2 wild-type exon 3 (blue droplets), partitions containing only positive transcripts for SMN1/SMN2 exon 7 (green droplets), and partitions containing doubly positive transcripts for both SMN1/SMN2 exon 7 and exon 3 (orange droplets). Thus, orange droplets represent exon 3 and exon 7 co-segregation signals due to full-length transcripts from both SMN genes. Blue and green dots are representative of truncated transcripts in which only exon 7 (VIC-green dots) or exon 3 (FAM-blue dots) are present. Template fragmentation is a random event that can occur during the droplet generation step. Note that, as already reported in the 1D-plot in Figure 1, the 2D-plot in Figure 2 also reveals two of VIC-positive green droplet populations. Those with a lower fU intensity are referred to the SMN2 transcript while the brighter ones are referred to the SMN1 transcript. Similarly, two populations of orange droplets, positive for co-segregation of exon 3 and exon 7 of both SMN genes are generated and can be correctly attributed to SMN1 or SMN2.

Figure 2: Representative 2-D plot of the fluorescence amplitude generated by QuantaSoft™ software of a healthy subject using the SMN-1 specific probe (VIC) in combination with the wild-type exon 3 probe (FAM).
The figure shows: i. a black cluster representing negative droplets; ii. two green clusters representing positive droplets for exon 7 only (SMN2 and SMN1 positioned closer and further away from the empty black droplets respectively); iii. a blue cluster representing droplets positive only for wild-type exon 3, iiii. two orange populations of droplets (VIC plus FAM) positive for both wild-type exon 3 and exon 7 that co-segregate within the full-length transcripts of SMN1 or SMN2. The most abundant cluster of orange droplets is representative of co-segregation of wild type exon 3 and SMN1 exon 7, whereas the poorest cluster of doubly positive droplets represents co-segregation of SMN2 exons 3 and 7. SMN EX3 WT+ = SMN exon 3 Wild Type positive droplets; SMN1 EX7+/EX3 WT+: SMN1 exon 7 and exon 3 Wild Type positive droplets; SMN2 EX7+/EX3 WT+: SMN2 exon 7 and exon 3 Wild Type positive droplets; SMN1 WT+: SMN1 exon 7 positive droplets; SMN2 EX7+: SMN2 positive droplets.
The positive events for each cluster reflected the amount of transcripts detected in the healthy control subject and, as expected, SMN1-derived mRNA was much more abundant than SMN2-derived mRNA.
Co-segregation study of exon 3 wt and mutant sequence with SMN1 and/or SMN2 transcripts
To unravel which SMN transcript (SMN1 or SMN2) carries the exon 3 deleterious point mutation in the analyzed subjects, a multiplex ddPCR assay was conducted performing simultaneous PCR amplification and fluorescence detection of SMN exon 7 and SMN exon 3. The overall results are shown in Figures 3 and 4. In both figures, panels A and D of the 2D plot show representative plots of the healthy control subject; panels B and E show plots of the healthy carrier and panels C and F show plots of the SMA patient. In detail, multiplex ddPCR were conducted using: i. The SMN1-specific probe plus either the wild-type exon 3-specific probe (panels A, B and C, Figure 3) or the mutant exon 3-specific probe (panels A, B and C, Figure 4); ii. The SMN2-specific probes plus either the wild-type exon 3-specific probe (panels D, E and F, Figure 3) or the mutant exon 3-specific probe (panels D, E and F, Figure 4). As previously specified, SMN1 or SMN2 positive droplet are represented as green plots of increasing fU on the x-axis of the graphs while the blue droplets are representative of the exon 3 signal of increasing fU on y-axis. Single positive droplets, as explained above, result from the fragmentation of transcript that occurred during the droplet generation process. Double positive droplets, typical of co-amplification of exon 3 and exon 7, are shown in orange. Black dots are always represented as empty droplets. Only the double positive orange droplets containing the full-length SMN transcript, including exon 3 and exon 7, were considered and counted in the analysis. In Figure 3, where the wild-type exon 3 probe was used in combination with exon 7 SMN1 (panels A, B, C) and SMN2 (panels D, E, F) probes, two clusters of orange droplets emerged, consistent with co-segregation of wild-type exon 3 with both SMN1 and SMN2 transcripts.

Figure 3: 2-D plots showing representative pattern results of positive droplets obtained after co-amplification with ddPCR of exon 7 and wildtype exon 3 of SMN1 and SMN2 transcripts, using SMN1 (panels A, B, C) and SMN2 (panels D, E, F) specific probes, respectively.

Figure 4: 2-D plots showing representative pattern results of positive droplets obtained after co-amplification with ddPCR of exon 7 and mutant exon 3 of SMN1 and SMN2 transcripts, using SMN1 (panels A, B, C) and SMN2 (panels D, E, F) specific probes, respectively.
Otherwise, using the exon 3 mutant probe, as shown in Figure 4, no mutated copies of exon 3 were found on either SMN1 or SMN2 templates in the healthy control subject. In the healthy carrier, a small number of mutated copies of exon 3 were found to segregate on the SMN1 transcript. Most of the signals derived from doubly positive droplets composed of SMN1 or SMN2 transcripts were characterized by a wild-type exon 3 profile. Interestingly, most of the full-length SMN1 transcripts carrying the deleterious exon 3 SNP were found in the SMA patient using both SMN1-specific and SMN2-specific probes (panels C and F, Figure 4). Remarkable is the switching of the position of the orange droplets in panels C and F of figure 4 that emphasize the co-segregation of exon 3 point-mutation with the SMN1 transcript. A very small amount of doubly positive orange droplets was detected with both SMN2 probes of exon 7 and mutant exon 3 in panel F of Figure 4 compared with the SMA patient analysis. However, considering the high abundance of the SMN2-derived transcript and the relative paucity of SMN1-derived transcript detectable for this patient we can assume some degree of cross hybridization of mutant exon 3 probe with the SMN2 transcript. In summary, the method we have developed is functional for the identification of point mutations located in SMN1 or SMN2 and could greatly simplify laboratory procedures for unambiguous assignment of SNPs to the gene and facilitate diagnosis for patients with SMA and healthy carriers at reproductive risk. This method could also potentially be applied to SNPs co-segregation studies for recessive diseases as discussed below.
Positive droplets, containing at least one copy of the amplified target, showed greater fluorescence than negative droplets. VIC-positive droplets are shown in green and are representative of SMN1 and SMN2 exon 7 transcripts; FAM-positive droplets are blue and relative to the exon 3. The doubly positive droplets, showed in orange, contained full-length transcripts, positive for both wild-type exon 3 and exon 7 of both SMN genes. Negative droplets (black dots) are separated from the others by a threshold (pink line) that defines positivity.
Representative plots of the healthy control subject are shown in panels A and D: no SNP co-segregation emerged from the analyses; plots B and E are relative to the healthy carrier and showed a small amount of SNPs of exon 3 co-segregating with the SMN1 transcript; plots C and F are relative to the SMA patient and showed the large majority of mutated copies of exon 3 co-segregating with the SMN1 transcript. It is interesting to remark the switching of droplet position using probes specific for SMN1 or SMN2 while keeping their fractional abundance constant.
Relative quantification of wild-type and mutant SMN transcripts using absolute positive droplet counts specific to exon 7 and exon 3.
The analytical step of a ddPCR-based assay involves setting a specific fluorescence threshold that separates the positive and negative partitions to estimate the steady state level of expression of each target gene. In this way, the absolute quantification and relative abundance of SMN1 and SMN2 transcripts as well as wild-type and mutated transcripts, can be done simultaneously in the absence of an internal standard calibrator.
Referring to the FAM-signal, specific for the characterization of exon 3 SNPs and co-segregation, two different PCR reactions were performed to identify the absolute number of wild-type and mutated exon 3 copies. Exon 3 copy number was calculated using two different strategies: (1) the presence of exon 3 in the same droplet with exon 7 SMN1 or exon 7 SMN2 derived from a full-length transcript (double positive FAM plus VIC signal) (2) the presence of single signals on FAM or VIC channels derived from template fragmentation that occurred during droplet generation. The proportions of SMN1 and SMN1 mutant transcripts were then estimated by calculating the ratio of SMN1 positive droplets (orange + green droplets) to the total number of SMN transcripts (SMN1+SMN2 orange + green droplets) and the ratio of SMN1 mutant transcripts to total SMN transcripts. The data are shown in Tables 2 and 3, respectively.
Absolute number of total SMN1 transcripts (column 2) and absolute number of total SMN transcripts (column 3) observed for each subject and used to calculate the percentage of SMN1 mRNA to total SMN transcripts (column 4).
Absolute number of mutant SMN1 exon 3 positive transcripts (column 2) and absolute number of SMN exon 7 total transcripts (column 3) observed for each subject and used to calculate the percentage of SMN1 mutant mRNA to total SMN exon 7 transcripts (column 4).
EX7= exon 7; EX3= exon 3
As we expected, the healthy subject and healthy carrier show a higher amount of SMN1 transcript (90% and 77%, respectively) than SMN2, consistent with data reported in the literature about insufficient levels of functional SMN transcript, including exon 7, produced by SMN2. In contrast, the SMA patient is characterized by very low levels of SMN1-derived transcript (6,3% in Table 2) approximately 16-fold lower than SMN2. This result cannot be justified by the presence of three copies of SMN2 and one copy of SMN1 genes, given the large difference in the efficiency of full-length RNA production by the 2 genes. Rather, as well documented by the results of our multiplex ddPCR assay, SMN1 of the SMA patient carries the deleterious point mutation in the exon 3 and this suggests a deleterious effect of the point mutation on the transcription process and/or transcript stability of SMN1 even before the generation of a truncated SMN protein. This concept is further supported by the results for the fraction of mutant-specific SMN1 transcript from the healthy carrier and SMA patient shown in Table 3.
Discussion
Here we describe a new method that provides for rapid assignment of coding point mutations, first identified by sequence, to SMN1 or SMN2 without the need for cloning of transcripts and screening of bacterial colonies or genespecific LR-PCR. Although most cases of SMA result from homozygous deletion of SMN1, the list of deleterious SMN1 point mutations is increasing. The analysis of SMN1 point mutations is not only useful for the diagnosis of those patients with heterozygous SMN1 deletion, who always have a copy or copies of SMN2, but will be useful for genetic counselling to their families, identification of healthy carrier relatives at reproductive risk, and prenatal diagnosis. This method combined the high performance of ddPCR in terms of target partitioning and quantification with the high sensitivity of TaqMan MGB probes to single SNPs within the specific target. The method we developed is not intended to find out new deleterious SMN1 mutations of a compound heterozygous SMA patient but aims to carry out a rapid and safe identification of the gene, SMN1 or SMN2, in which a previously identified coding or splicing mutation segregates, improving clinical outcome.
ddPCR is a novel technology based on random template partitioning and the ability to perform multiple independent PCR amplifications in water-in-oil droplet emulsions. In each droplet, the absolute number of target nucleic acid molecules can be measured using the binomial Poisson statistics and defined as the absolute concentration directly from the number of positive droplets to the total. Due to its high sensitivity, ddPCR is often used in pathological conditions such as somatic mosaicisms, CNV, and for the identification of rare allelic variants and non-abundant RNA transcript. TaqMan MGB probes are usually used in multiplex Real-Time PCR assays for relative copy number quantification of SMN1 and SMN2 (Lonigro et al. unpublished data) for identification of SMA patients and SMA healthy carriers. However, Real- Time based assays and analysis software allow visualization and quantification of the fluorescence emitted by TaqMan MGB probes hybridized to the specific target, while any signals due to non-specific probe cross-hybridization, such as SMN1 probe on SMN2 and vice versa, are subtracted as background due to the low emitted energy. Similar multiplex assays performed with ddPCR technology allow visualization and consequent analysis of all droplets positive for a fluorescent probe. In the analysis we performed we obtained simultaneous visualization of droplets positive for both, SMN1 and SMN2 in different areas of the 2D graph. Precisely, highly specific probe/target interactions (SMN1 probe on SMN1 and SMN2 probe on SMN2) are displayed in the highest energy-emitting area of the 2D plot, whereas non-specific probe/target interactions (SMN1 probe on SMN2 and SMN2 probe on SMN1) are displayed in the lowest energy-emitting area of the plot. Simultaneous visualization of the SMN1 and SMN2 positive droplets, using both the SMN1- and SMN2- specific TaqMan MGB probe, allows a simple test based on multiplex ddPCR co-segregation to assign the deleterious SNP(s) identified in the coding sequence of the SMN genes to either SMN1 or SMN2 undoubtedly. In our case, the SNP identified in exon 3 of the SMN genes of a subject with SMA and an unrelated SMA carrier is known to be deleterious because it introduces a stop codon leading to a truncated SMN protein. This point mutation, already known to be in SMN1, demonstrate the validity of the method we developed, based on an unconventional use of ddPCR technology and TaqMan MGB chemistry, since it can be undoubtedly assigned to SMN1. This method avoids tedious and time-consuming procedures connected to transcripts cloning and bacterial colony screening or to LR-PCR procedures. Moreover, the manipulation of bacterial and human biological materials needs specific environments and devices. In addition, our method allows to quantify the relative abundance of SMN1 and SMN2 full-length transcripts confirming the well-known difference in the transcriptional activity of the two genes. Surprisingly, the relative abundance of the SMN1-derived transcript with the point mutation in exon 3 was very low in both in the SMA patient and the healthy carrier. This observation might suggest that the point mutation has a deleterious effect on the transcriptional activity of the gene or the stability of the transcript even before the generation of a truncated protein. This observation is a novelty never reported before as result of the already known point mutation in exon 3 of SMN1. More generally, we cannot exclude that such a deleterious effect on mRNA transcription end/or stability may also occur in the case of other point mutations, perhaps conservative on proteins, which should always be investigated since the recent observation that intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy number.
Conclusion
Finally, this method has the potential to become a complementary diagnostic tool in detecting mono or biallelic segregation of deleterious point mutations in recessive inherited diseases such as some of the results obtained through NGS (Next Generation Sequencing) based tests, without the need to analyze parents or other relatives of the patient. The segregation study of healthy parents and other relatives of the affected subjects is the elective procedure for definitive diagnosis of recessive inherited diseases and identification of healthy carrier/s. However, other subjects within the family of a patient are not always available and the method we have developed can overcame this unavailability and still allow the diagnosis to be made.
References
- Zapletalova E, Hedvicakova P, Kozak L, et al. Analysis of point mutations in the SMN1 gene in SMA patients bearing a single SMN1 copy. Neuromuscul Disord. 2007;17(6):476-81.
- Melki J, Lefebvre S, Burglen L, et al. De novo and inherited deletions of the 5q13 region in spinal muscular atrophies. Sci. 1994;264(5164):1474-7.
- Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. J Eur Hum Genet. 2004;12(12):1015-23.
- Lefebvre S, Bürglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155-65.
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30(4):377-84.
- Parsons DW, McAndrew PE, Monani UR, et al. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet. 1996;5(11):1727-32.
- Passon N, Pozzo F, Molinis C, et al. A simple multiplex real-time PCR methodology for the SMN1 gene copy number quantification. Genet Test Mol Biomark. 2009;13(1):37-42.
- Wirth B, Herz M, Wetter A, et al. Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. J Am Hum Genet. 1999;64(5):1340-56.
- Stuppia L, Antonucci I, Palka G, et al. Use of the MLPA assay in the molecular diagnosis of gene copy number alterations in human genetic diseases. J Int Mol sci. 2012;13(3):3245-76.
- Tomaszewicz K, Kang P, Wu BL. Detection of homozygous and heterozygous SMN deletions of spinal muscular atrophy in a single assay with multiplex ligation-dependent probe amplification. J Peking Univ Health Sci. 2005;37(1):55-7.
- Passon N, de Wittenau GD, Jurman I, et al. Quick MLPA test for quantification of SMN1 and SMN2 copy numbers. Mol Cell Probes. 2010;24(5):310-4.
- Cusco I, Barceló MJ, Del Río E, et al. Detection of novel mutations in the SMN tudor domain in type I SMA patients. Neuro. 2004;63(1):146-9.
- Clermont O, Burlet P, Benit P, et al. Molecular analysis of SMA patients without homozygous SMN1 deletions using a new strategy for identification of SMN1 subtle mutations. Hum Mutat. 2004;24(5):417-27.
- Kubo Y, Nishio H, Saito K. A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J Hum Genet. 2015;60(5):233-9.
- Alías L, Bernal S, Fuentes-Prior P, et al. Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum genet. 2009;125(1):29-39.
- Li W. Structural and Functional Consequences of the SMA-Linked Missense Mutations of the Survival Motor Neuron Protein: A Brief Update. InNovel Aspects on Motor Neuron Disease. 2018.
- Vogelstein B, Kinzler KW. Digital pcr. PNAS. 1999;96(16):9236-41.
- Bizouarn F. Digital PCR: Improving Nucleic Acid Quantification: Precision, Accuracy, and Sensitivity Are Among the Benefits Reported by Researchers. Genet Eng Biotechnol News. 2012;32(9):32-3.
- Huggett JF, Cowen S, Foy CA. Considerations for digital PCR as an accurate molecular diagnostic tool. Clin Chem. 2015;61(1):79-88.
- Debski PR, Garstecki P. Designing and interpretation of digital assays: Concentration of target in the sample and in the source of sample. BDQ. 2016;10:24-30.
- Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. PNAS. 1999;96(20):11167-72.
- Hindson BJ, Ness KD, Masquelier DA, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604-10.
- Mazaika E, Homsy J. Digital droplet PCR: CNV analysis and other applications. Curr Protoc Hum Genet. 2014;82(1):7-24.
- Baker M. Digital PCR hits its stride. Nat Methods. 2012;9(6):541-4.
- Morley AA. Digital PCR: A brief history. BDQ. 2014;1(1):1-2.
- Olmedillas-López S, García-Arranz M, García-Olmo D. Current and emerging applications of droplet digital PCR in oncology. Mol Diagn Ther. 2017;21(5):493-510.
- Nadauld L, Regan JF, Miotke L, et al. Quantitative and sensitive detection of cancer genome amplifications from formalin fixed paraffin embedded tumors with droplet digital PCR. Transl Med. 2012;2(2).
- Brichta L, Garbes L, Jedrzejowska M, Grellscheid SN, Holker I, Zimmermann K, Wirth B. Nonsense-mediated messenger RNA decay of survival motor neuron 1 causes spinal muscular atrophy. Hum Genet. 2008;123(2):141-53.
- How TaqMan SNP Genotyping Assays Work - US. Available: //www.thermofisher.com/us/en/home/life-science/pcr/real-time-pcr/real-time-pcr-learning-center/genotyping-analysis-real-time-pcr-information/how-taqman-snp-genotyping-assays-work.html
- Leiden Muscular Dystrophy Pages. Available: https://www.dmd.nl/
- Wijaya YO, Rohmah MA, Niba ET, et al. Phenotypes of SMA patients retaining SMN1 with intragenic mutation. Brain Dev. 2021;43(7):745-58.
- Park S, Lee H, Shin S, et al. Analytical validation of the droplet digital PCR assay for diagnosis of spinal muscular atrophy. Clin Chim Acta. 2020;510:787-9.
- Vidal-Folch N, Gavrilov D, Raymond K, et al. Multiplex droplet digital PCR method applicable to newborn screening, carrier status, and assessment of spinal muscular atrophy. Clin Chem. 2018;64(12):1753-61.
- Corbisier P, Pinheiro L, Mazoua S, et al. DNA copy number concentration measured by digital and droplet digital quantitative PCR using certified reference materials. Anal Bioanal Chem. 2015;407(7):1831-40.
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref
Indexed at, Google Scholar, Cross Ref