- Journal of RNA and Genomics (2008) Short Report
Native microRNA loop sequences can improve short hairpin RNA processing for virus gene silencing in animal cells
| Tracey M Hinton1,2*, Terry G Wise1, Pauline A Cottee1 and Timothy J Doran1,2 1CSIRO Livestock Industries, Australian Animal Health Laboratory, Geelong 3220, Australia 2Cooperative Research Centre for the Australian Poultry Industry, Armidale, NSW, 2315, Australia. |
| *Correspondence to: Tracey Hinton, Email: Tracey.Hinton@csiro.au, Tel: +61 352275746, Fax: +61 352275555 |
| Received 19 March 2008; Revised 06 May 2008; Accepted 14 May 2008; Published online 27 May 2008 |
| © Copyright The Authors: This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/). This license permits noncommercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Abstract
Introduction of small interfering RNAs (siRNAs) into cells results in transitory silencing of target genes with complementary sequence. Incorporating siRNAs into short-hairpin RNAs (shRNAs) or microRNA-adapted shRNAs (shRNAmir) is a popular tool for targeted gene silencing. shRNAmirs mimicking endogenous premicroRNAs (unprocessed hairpin microRNAs) are more difficult to design and result in longer RNA molecules. The use of microRNA (miRNA) loop sequences in shRNAs as an alternative to an entire premicroRNA structure on silencing efficiency has not been studied extensively. This report shows that loop sequences derived from native miRNAs improves the efficiency of silencing due to the processing of the shRNAs into mature siRNAs.
Keywords |
| microRNA; RNA interference; short hairpin RNA, influenza |
Introduction |
| Animal cells use RNA interference (RNAi) as a natural mechanism to regulate gene expression through the use of miRNAs. miRNAs are transcribed from the genome as primary miRNA transcripts (pri-miRNA), which are processed by Drosha into precursor miRNA hairpins (premiRNA: Lee et al, 2003). Exportin-5 transports the hairpin from the nucleus into the cytoplasm (Yi et al, 2003) where the loop of the hairpin precursor is removed by the ribonuclease III enzyme Dicer, leaving the mature 22-25 nt double stranded miRNA (Zamore et al, 2000). The miRNA guide strand loads into the RNA induced silencing complex (RISC) which directs it to the complementary messenger RNA (mRNA) resulting in cleavage of the target mRNA (Hammond et al, 2000). Thousands of miRNAs are predicted to be present in every cell and many of these miRNAs are highly regulated to be tissue or cell cycle specific (Thomson et al, 2006; Obernosterer et al, 2006). The cellular RNAi mechanism has been successfully adapted to specifically silence genes of interest including viral and endogenous genes as reviewed in Pushparaj PN and Melendez AJ (2006). |
| One method to artificially induce RNAi induced gene silencing is to express shRNAs. shRNAs consist of a siRNA target sense sequence acting as the 5’ stem, a spacer sequence which forms the loop and the anti-sense sequence forming the 3’ stem. An alternative method is to mimic naturally occurring pri-miRNA structures shRNAmirs (Boden et al, 2004; Siolas et al, 2005). Once transcribed from an expression vector these molecules enter the RNAi pathway at the Drosha step for shRNAmirs or the Dicer step for shRNAs, to be cleaved into siRNAs (Henry et al, 2006; Gou et al, 2007). |
| It has been shown that the shRNA loop sequence is critical for efficient mRNA silencing as the majority of the processing by Dicer occurs near the loop (McManus et al, 2002). Initial shRNA expression experiments showed that a 19 nt siRNA sequence and a 9 nt spacer was the most efficient and this structure has become the standard for shRNA design (Brummelkamp et al, 2002). The use of endogenous miRNA loop sequences to improve shRNA silencing has not been extensively investigated. To determine whether shRNA silencing of viral genes could be improved by the use of miRNA loop sequences, shRNAs targeting influenza A/PR/8/34 (PR8) strain Nuclear Protein (NP) mRNA and chicken anaemia virus (CAV) mRNA were designed. These shRNAs contain 19 nt siRNA target sequences with loop sequences derived from one of three native miRNAs (chicken miR17, miR30a and human miR30a) known to express highly in most cell types. Silencing was compared to the highly efficient 9 nt loop sequence described by Brummelkamp et al, (2002). |
Materials and Methods |
shRNA loop design and plasmid constructs |
| shRNAs targeting chicken anaemia virus (CAV) mRNA have previously been assayed for silencing against peGFPCAV (Hinton TM and Doran TJ, in press). To produce pEGFP-NP, a pGEMTeasy plasmid containing a 180 bp fragment of NP was digested with NotI. The NP fragment was gel purified and ligated into the similarly digested pEGFP-C (a gift from David Cummins, CSIRO Livestock Industries, Australia). The siRNA sequence targeting NP was obtained from Ge et al, (2003). NP shRNA molecules were designed with either the 9 nt hairpin loop sequence of Brummelkamp et al, (2002) or the microRNA loop sequences from human miR30a (miRBase ref: MI0000088), chicken miR30a (miRBase ref: MI0001204) and chicken miR17 (miRBase ref: MI0001184) obtained from miRBase (http://microrna.sanger.ac.uk/sequences). Complementary oligonucleotides were annealed and ligated into pchU6-4 as described previously (Hinton TM and Doran TJ, in press). The forward oligonucleotide sequences used are shown in Table 1 and were obtained from Geneworks (Australia). The resulting Influenza PR8 NP shRNA constructs have been designated pshNP-OL, pshNP-miR17, pshNP-miR30agga and pshNP-miR30ahsp. The CAV shRNA constructs have been named pshVP2/3- 1-OL, pshVP2/3-1-miR17, pshVP2/3-1-miR30agga, pshVP2/3-1-miR30ahsp, pshVP2/3-3-OL, pshVP2/3-3- miR17, pshVP2/3-3-miR30agga and pshVP2/3-3- miR30ahsp. All constructs were sequenced by Micromon DNA sequencing facility (Monash University, Australia). |
Cells and virus |
| Chicken fibroblast cells (DF1: ATCC No. CRL-12203) were grown in DMEM and Madin-Darby canine kidney cells (MDCK: ATCC No. CCL-34) were grown in EMEM, both were supplemented with 10% foetal bovine serum, 2 mM glutamine, 10 mM Hepes, 1.5 g/l sodium bicarbonate, 4.5g/l glucose, 0.01% (w/v) penicillin and 0.01% (w/v) streptomycin at 37ºC with 5% (v/v) CO2 and sub-cultured twice weekly. |
| Influenza A/PR/8/34 (PR8) strain virus stock was produced by limiting dilution passage in the allantoic cavity of 10 day old embryonated chicken eggs at 34°C for 48-72 hr. Virus was passaged three times. |
EGFP-fusion silencing |
| DF1 cells were seeded at 1.5x105 cells in 24-well tissue culture plates in duplicate and grown overnight at 37ºC with 5% (v/v) CO2. Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen, USA) as per manufacturer’s instructions. Briefly, 1 Ng of pEGFP-NP or pEGFP-CAV and 1 Ng of the relevant shRNA plasmid were mixed with 2 Nl of Lipofectamine 2000 both diluted in 100 Nl OPTI-MEM (Invitrogen, USA) and incubated at room temperature for 20 min. The DNA:lipofectamine mix was added to cells and incubated for 4 hr. Cell media was replaced and incubated for 72 hr. Cells were washed twice with PBS, trypsinised and washed twice with FACS wash (PBS with 1% (v/v) FBS). Cells were subjected to flow cytometry and EGFP silencing was analysed as a percentage of the non-silencing shRNA mean EGFP (measured on FITC wavelength) fluorescence. |
Influenza A-PR8 silencing |
| MDCK cells were transfected using Amaxa nucleofector electroporation (Amaxa Biosystems, Germany). Briefly 1.5x106 MDCK cells were pelleted and resuspended in 100 Nl of nucleofector T solution with DNA and electroporated with program T20. Cells were diluted with 500 μl of prewarmed growth media, aliquoted into 6 wells of a 24-well culture plate and incubated overnight at 37ºC with 5% (v/v) CO2. Influenza A PR8 virus was serially diluted in viral growth media (VGM, with 0.3% (v/v) BSA, 5 Ng/ml trypsin and lacking FCS) and cells were infected at multiplicities of infection (MOI) of 0.01, 0.001, 0.0001 in duplicate and were incubated at 37ºC for 1 hr, virus was replaced with VGM and incubated for 48 hr. Supernatant was taken and used in a haemagglutination assay according to the OIE Manual. Briefly, this involved serial two-fold dilutions of virus in PBS these were mixed with an equal volume of a 0.5% (v/v) chicken erythrocyte suspension. After 1 hr incubation at room temperature, the HA titer was estimated by the highest dilution with hemagglutination. |
RNA isolation and northern blotting |
| DF1 cells were seeded and grown to 80% confluency in 25 cm tissue culture flasks (Nunc, USA). Plasmids were transfected into the DF1 cells using Lipofectamine 2000. Briefly, 12 Ng of the relevant shRNA plasmid was mixed with 20 μl of Lipofectamine 2000 and transfected as described previously. RNA (<200 nt) was purified from transfected cell cultures using mirVanaTM miRNA isolation kits (Ambion, Austin USA) and concentrated using Millipore microcon centrifugal filters (YM-30; Millipore, USA). Approximately 1 Ng of low molecular weight RNA was resolved on a 7M Urea-15% (w/v) polyacrylamide gel and transferred to a positively charged membrane (Hybond plus, Amersham Biosciences, USA) using a Trans-blot semi-dry transfer cell (BioRad, USA). The efficiency of each hairpin expression and processing was determined using the NP-Locked Nucleic Acid (LNA) probe (5'-CTCCGAAGAAATAAGATCC-3'; Sigma- Proligo, USA) that was end-labeled with [32SP] dATP using 10 U of OptiKinase (USB, USA). Underlined nucleotides indicate the locked bases. Hybridization was conducted overnight at 42oC in 50% (v/v) formamide, 0.5% (w/v) SDS, 5x SSPE, 5x Denhardts solution and 100 μg/ml denatured herring sperm DNA (Roche, USA). The membrane was washed 3 times in 2x SSC, 0.1% (w/v) SDS at 42oC prior to overnight autoradiographic exposure. The size of the resolved RNA was determined by comparison with AmbionTM Decade markers (Ambion). |
 |
| Table 1: Forward shRNA oligonuleotide sequences |
Results |
Influenza PR8 NP shRNA loop sequences and plasmid constructs |
| The loop sequences used in this study were obtained from either Brummelkamp et al. (2002) or from miRBase. We chose the human miR30a loop sequence (miR30ahsp) as miR30a based shRNAmirs express high levels of siRNAs (Boden, et al, 2004). For silencing chicken pathogens, the chicken miR30a (miRgga30a) loop sequence was also selected, as it contains two nucleotide differences to the human version. The chicken miR17 (miR17) loop sequence was selected as the native miRNA is expressed at high levels in all chicken cell types (ICGSC, 2004). A siRNA targeting Influenza NP was adapted to shRNAs containing one of the four loop sequences (Table 1). The resulting plasmids are referred to as pshNP-OL, pshNPmiR17, pshNP-miR30agga and pshNP-miR30ahsp. Figure 1A describes the predicted structures and UG values of the original shRNA structure of Brummelkamp et al, (2002) and the native microRNAs, whilst the predicted structures and UG values of the NP hairpins are shown in Figure 1B. It should be noted that 4 of the nucleotides in the Brummelkamp shRNA loop sequence are predicted to base-pair. The miR30a shRNA structure predictions and UG values closely resemble those of the native miRNAs, missing one 2 nt bulge, whilst the miR17 shRNA loop matches the structure predicted for the miRNA, however the stem appears quite different as it does not contain the multiple bulges. All shRNAs were under the control of the chicken U6-4 promoter (chU6-4), along with the nonsilencing (pshNS) and positive (pshEGFP, Table 1) control described previously (Wise et al, 2007). |
Chicken miR17 loop sequence decreases the ability of the shRNA to silence EGFP-fusion expression, by inhibiting processing of the hairpin to mature siRNAs |
| Prior to virus silencing experiments, each NP shRNA vector was assayed for activity against EGFP-NP fusion mRNA in the chicken fibroblast cell line DF1. pshNP-OL was highly active at silencing pEGFP-NP mRNA (Figure 2A). pshNP-miR30agga and pshNP-miR30ahsp showed a marginal increase in silencing pEGFP-NP compared to pshNP-OL (Figure 2A). Inclusion of the chicken miR17 loop sequence resulted in a 3-fold decrease in EGFP-NP mRNA silencing (Figure 2A). |
| To determine why pshNP-miR17 was less active than other constructs, small RNAs were isolated from transfected DF1 cells and analysed by northern blot (Figure 2B). This method detects both the hairpin structure and the mature siRNA using a probe directed at the NP siRNA sequence. Hairpin and mature siRNA were detected for pshNP-OL and both miR30a constructs (Figure 2B). The NP miR17 hairpin (pshNP-miR17) was detected, but no mature sequence was observed (Figure 2B). The northern blot shows a high level of unprocessed hairpin present from all vectors compared to the level of mature siRNAs observed (Figure 2B). A higher concentration of mature siRNA was present from the miR30a constructs. No bands were observed in the untransfected or non-silencing control (Figure 2B). |
 |
| Figure 1: Schematic representation of influenza H1 NP targeting shRNAs with different microRNA loops. A: Native pre-miRNAs. The red letters are the siRNA sequences, black letters indicate extra miRNA stem sequences, blue letters indicate the loop sequence used and underlined letters indicate loop bases not present in the shRNA constructs. B: NP targeting shRNAs with microRNA loops predicted through mFOLD. The red letters are the siRNA sequences and the blue letters indicate the loop sequences used. |
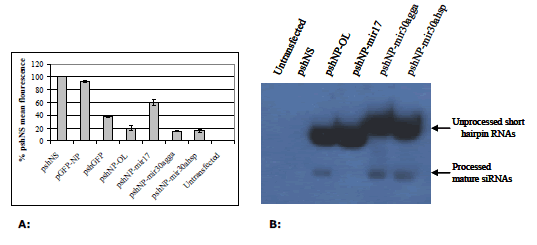 |
| Figure 2: A: Silencing of EGFP-NP fusion mRNA by shRNAs in DF1 cells. DF1 cells were co-transfected with 1 μg each of the relevant vectors as indicated in X axis and 1μg of pEGFP-NP where required for 72 hr. Cells were then assayed by flow cytometry and analysed in Microsoft Excel. Values are shown as percentages of the negative control shRNA (shNS), as the mean of three separate experiments in duplicate ± standard deviation. B: Verification of shRNA and siRNA expression by northern blot. Northern blot of NP targeted shRNA molecules. |
shRNA silencing of viral RNA mimics the EGFP-fusion assay |
| The shRNA constructs were assayed for the ability to silence Influenza A strain PR8 in MDCK cells. The haemagglutination assay showed consistent results to the EGFP reporter assay with highly efficient knockdown of virus replication observed with the original loop construct (Figure 3). The miR30a loop sequences increased the silencing ability of the NP siRNA at the highest concentration of virus, with the chicken miR30a loop giving the best knockdown (Figure 3). As expected, pshNP-miR17 was unable to inhibit viral replication efficiently. |
Different loop sequences do not improve less efficient siRNA molecules |
| To determine if the loop sequence affected the silencing ability of other shRNA sequences, vectors expressing shRNAs with the four different loop sequences targeting chicken anaemia virus mRNA were produced. Two shRNA sequences were analysed, one highly active against EGFP-CAV fusion mRNA (pshVP2/3-1) and another less active sequence (pshVP2/3-3; Hinton TM and Doran TJ, in press). The resulting constructs are referred to as pshVP2/3-1-OL, pshVP2/3-1-miR17, pshVP2/3-1- miR30agga, pshVP2/3-1-miR30ahsp, pshVP2/3-3-OL, pshVP2/3-3-miR17, pshVP2/3-3-miR30agga and pshVP2/3-3-miR30ahsp. The same non-silencing control (pshNS) and EGFP targeted shRNA (pshGFP) were utilised. Similar silencing results to those observed with the NP shRNAs were obtained for the CAV shRNA contructs in the DF1 GFP reporter assay (Figure 4). The shRNAs containing the miR17 loop sequence were less active, whilst the chicken miR30a loop shRNAs was the most efficient (Figure 4, A and B). Interestingly, the different loop sequences examined were unable to improve the activity of shVP2/3-3, indicating that whilst a loop sequence can improve hairpin processing, they are unable to improve the ability of a siRNA to silence the target gene (Figure 4B). |
Discussion |
| shRNAmirs are processed by Drosha into shRNAs. shRNAs are then recognized and processed into siRNAs by Dicer (Paddison et al, 2002). Initially shRNAmirs appeared to produce more mature siRNAs resulting in better silencing (Boden et al, 2004; Silva et al, 2005). However, McManus et al, (2002) has shown that the loop sequence is the most critical region in recognition of the shRNA for processing. To determine if a native miRNA loop sequence could improve shRNA processing and increase silencing, a comparison of shRNAs containing either the commonly used 9nt loop sequence from (Brummelkamp et al, 2002) and three endogenous miRNA loop sequences was performed. |
| The miR17 loop sequence used in this study was selected based on the unpaired nucleotides in the predicted miRNA structure. Subsequent analysis of chicken miR17 sequence revealed four additional bases extend 3’ from the loop sequence, two of which are base-paired with the siRNA (miRBase, Figure 1A). Chicken miR17 also contains several bulges in the stem sequence not present in the shRNAs, resulting in the shRNA predicted structure looking different (Figure 1B). Little is known about the shRNA structure requirement for Dicer cleavage. McManus et al, (2002) has demonstrated that the processing of shRNAmirs was sensitive to structural modifications including bulge position and loop sequence. The absence of the bulges in miR17 stem may have contributed to the inefficient shRNA processing. It would be interesting to test a NP-miR17 based shRNAmir with the bulges and paired loop sequence incorporated to see if processing is restored. The miR30a shRNA constructs, despite missing a 2nt bulge in the stem mimic the endogenous miRNA more accurately resulting in correct recognition and processing (Figure 1B). Therefore it appears important to choose loop sequences that will result in an shRNA that closely mimics the endogenous miRNA structure for improved silencing. |
| Although chicken miR17 is highly and ubiquitously expressed in chicken cells (ICGSC, 2004), it is transcribed from the miR17-92 miRNA cluster which produces 6 miRNAs. One of these, miR18a is known to require a cellular protein, the nucleo-cytoplasmic shuttling protein hnRNP A1, to be processed (Guil and Caceres, 2007). No similar factor has been determined for miR17, however this could explain the inefficiency of miR17 shRNA’s processing. Understanding the factors that regulate processing of shRNAs containing miRNA loop sequences would be advantageous in many circumstances. It could lead to targeted expression of the shRNA within cells or tissues, however, further studies are required in this area. |
| A recent paper by Liu, et al (2008) used the miR17-92 cluster as a backbone for expressing multiple siRNAs targeting HIV. The HIV siRNA expressed from the miR17 position gave minimal silencing as a single hairpin transcript, however silencing efficiency increased when the siRNA was present in a polycistronic transcript. The difference in silencing was thought to be due to misfolding of the hairpin and decreased processing to mature siRNA when present singly. Therefore the use of loop sequences from polycistronic miRNAs may be problematic. |
Conclusions |
| This study indicates that endogenous miRNA loop sequences that are derived from the animal host of the target virus can increase the efficiency of mature siRNA production. However the choice of the loop sequences requires careful consideration of the miRNA it is obtained from. In the future, understanding the efficiency and specificity of miRNA loop sequences may prove useful for delivering tissue targeted gene silencing in either single or multiple expression constructs. |
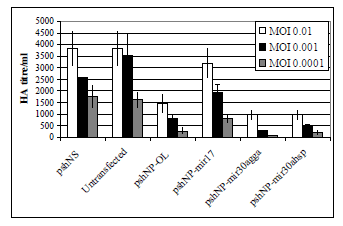 |
| Figure 3: Silencing of influenza A PR8 by shRNAs in MDCK cells. MDCK were electroporated with 2.5 μg of DNA in nucleofector solution T with Amaxa program T20. Plasmids used are indicated in X axis. Transfected cells were incubated for 24 h then infected with Influenza A PR8 virus for 48 h. Supernatants were assayed for Influenza A virus by HA assay. Graph depicts two separate experiments in duplicate ± SEM. |
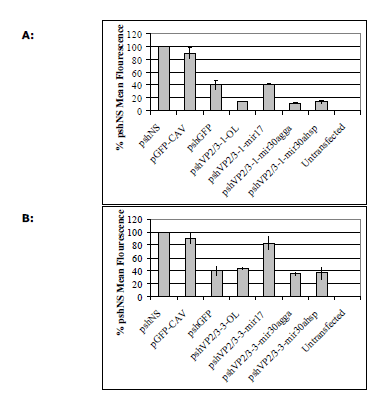 |
| Figure 4: Silencing of targeted EGFP-CAV fusion mRNA by shRNAs in DF1 cells. A. DF1 cells were co-transfected with 1 μg each of the relevant pshVP2/3-1 vectors as indicated in X axis and 1μg of pEGFP-CAV where required for 72h. B. DF1 cells were cotransfected with 1 μg each of the relevant pshVP2/3-3 vectors as indicated in X axis and 1μg of pEGFP-CAV for 72 hr. Cells were then assayed by flow cytometry and analysed in Microsoft Excel. Values are shown as percentages of the non-silencing control shRNA (shNS), as the mean of three separate experiments in duplicate ± standard deviation. |
Acknowledgements |
| This work was partly conducted within the Australian Poultry co-operative research centre, established and supported under the Australian Government’s co-operative research centres program and CSIRO-Livestock Industries. We would like to acknowledge D Cummins for his kind donation of the pEGFP-C plasmid. |
Competing Interests |
| None declared. |
List of Abbreviations |
| CAV: Chicken anaemia virus |
| GFP: Green fluorescent protein |
| gga: Gallus gallus - chicken |
| hsp: Homo sapien - human |
| miRNA: MicroRNA |
| NP: Nuclear protein from influenza |
| RISC: RNA induced silencing complex |
| siRNA: Small interfering RNA |
| shRNA: Short hairpin RNA |
References
|