- Journal of RNA and Genomics (2012) Volume 8, Issue 1
Knockdown of AMP-activated protein kinase alpha 1 and alpha 2 catalytic subunits
| Larissa Tangeman, Christopher N Wyatt* and Thomas L Brown* Program in Microbiology and Immunology and Department of Neuroscience, Cell Biology and Physiology, Wright State University Boonshoft School of Medicine, Dayton, Ohio 45435, USA |
| Corresponding Author: Thomas Brown, Email: thomas.L.brown@wright.edu, or Christopher Wyatt, Email: christopher. wyatt@wright.edu, Tel: +937 7753809, Fax: +937 7753391 |
| Received 08 May 2012; Revised 25 September 2012; Accepted 03 October 2012; Published 04 October 2012 |
| © Copyright The Author(s): Published by Library Publishing Media. This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.5). This license permits non-commercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Abstract
AMP-activated protein kinase (AMPK) is a master metabolic regulator that responds to the AMP: ATP ratio and promotes ATP production when the cell is low on energy. There are two isoforms of the catalytic alpha subunit, AMPKα1 and AMPKα2. Here, we describe the production of a small interfering RNA (siRNA) and a short hairpin RNA (shRNA) targeting both catalytic isoforms of AMPK in human, mouse, and rat. Multiple loop sequences were tested to generate the most effective shRNA. The shRNA causes significant knockdown of both isoforms of AMPKα in mouse and human cells. The shRNA effectively knocked down AMPKα1 and AMPKα2 protein levels, compared to a five basepair mismatch-control shRNA in mouse fibroblast NIH3T3 cells and significantly knocked down AMPKα1 (63%) and AMPKα2 (72%) levels compared to control in human embryonic kidney cells, HEK293s. The shRNA also causes a significant reduction in AMPK activity, measured as phosphorylation of acetyl-CoA carboxylase (ACC), a direct phosphorylation target. While the protein levels of total ACC remained the same between the AMPKα1and α2 shRNA and control shRNAtreated cells, there was a 41% reduction in phospho-ACC protein levels. The generation of this AMPKα1and α2 shRNA can be used to stably knock down protein levels and activity of both catalytic isoforms of AMPK in different species to assess function.
Keywords |
| AMPK, PRKAA, AMPKα1, AMPKα2, shRNA, siRNA |
Introduction |
| Regulating energy levels is an essential process that occurs in all living organisms. AMPK (5′-adenosine monophosphateactivated protein kinase), also known as protein kinase, AMP-activated (PRKAA), is an enzyme that is conserved from yeast to humans and has important roles in sensing the energy status of the cell to maintain homeostasis (Dale et al, 1995; Horman et al, 2002; Zhou et al, 2009; Viollet et al, 2009; Hardie, 2011). AMPK has been linked to numerous disease states, including metabolic disorders and cancer (Carling, 2005; Steinberg et al, 2009). Functional AMPK is a heterotrimer that acts to regulate metabolism and is composed of three subunits, the catalytic alpha subunit and the regulatory beta and gamma subunits (Viollet et al, 2009). There are two isoforms of the AMPK alpha subunit, AMPKα1 and AMPKα2, and each isoform has been shown to have overlapping as well as distinct functions depending on the cell type (Viollet et al, 2003; Neurath et al, 2006; Hardie, 2007; Viollet et al, 2009; Hawley et al, 2010). |
| AMPK acts as a metabolic master switch regulating several intracellular systems including glucose uptake and β-oxidation of fatty acids. AMPK inhibits fatty acid synthesis by phosphorylating acetyl-CoA carboxylase (ACC) (Park et al, 2002). In addition, AMPK has been reported to couple hypoxic inhibition of mitochondrial oxidative phosphorylation to carotid body type I cell excitation (Evans et al, 2005). The energy-sensing capability of AMPK can be attributed to its ability to detect and react to fluctuations in the AMP: ATP ratio. Stressors that deplete ATP in the cell result in an elevated AMP: ATP ratio, triggering AMPK phosphorylation and activation. Activated AMPK promotes ATP production and inhibits anabolic pathways that utilize ATP. |
| While it is clear that AMPK is a major regulator of cellular metabolism, there remain numerous unanswered questions about its function. Multiple strategies to manipulate AMPK activity have been developed, such as the use of the activating drug AICAR (5-aminoimidazole-4-carboxyamide ribonuclesoide) or the AMPK inhibitor, Compound C, but these pharmacological agents have been shown to alter other processes in the cell as well (Guigas et al, 2007; Emerling et al, 2009). The generation of AMPKα1 (a1-/-) and α2 (a2-/-) knockout mice has established critical roles for AMPKα1 and AMPKα2 in the regulation of energy metabolism and oxygen sensing (Viollet et al, 2003; Xing et al, 2003; Jorgensen et al, 2005; Viollet et al, 2009). Double knockout of the AMPKα1 and AMPKα2 isoforms, simultaneously, results in lethality at embryonic day 10.5 (Viollet et al, 2009). Therefore, an alternative model system is needed in order to study functions that can be performed by both isoforms of AMPKa. |
| RNA interference (RNAi) is a process by which doublestranded RNA (dsRNA) molecules can regulate gene expression. Small interfering RNAs (siRNAs), are 21-nucleotide dsRNA molecules that are complementary to a mRNA transcript (Fire et al, 1998). When siRNA is introduced into a cell, it can bind the complementary mRNA resulting in destruction of the mRNA transcript, thus preventing translation into protein (Elbashir et al, 2001; Kim et al, 2008; Carthew et al, 2009). Short hairpin RNAs (shRNAs) are siRNA molecules that have been modified to include a stem-loop-stem structure. They are transcribed from DNA templates such as plasmids, and this allows their expression to be stable and heritable in cell culture (Brummelkamp et al, 2002a; Brummelkamp et al, 2002b; Harboth et al, 2003). In addition, shRNAs can be used to generate transgenic animal models with knockdown of target proteins as an alternative to classic transgenic animal knockout models (Herold et al, 2008; Hitz et al, 2009). |
| In this study, we demonstrate the production of a novel shRNA that simultaneously targets both AMPKα1 and α2 isoforms. This shRNA is 100% complementary to a nucleotide sequence conserved in the human, mouse, and rat forms of the AMPKα1 and AMPKα2 mRNA and causes a significant reduction in AMPKα1 and α2 protein levels. Knockdown of AMPKα1 and α2 also reduces phosphorylation of ACC, a direct target of AMPK. This new shRNA will provide a useful new weapon to attack and analyze the numerous functional roles of AMPKα1 and α2. |
Materials and Methods |
| Cloning and cell culture |
| RNA oligonucleotides for siRNA experiments and DNA oligonucleotides for shRNA experiments were synthesized by Integrated DNA Technology (Coralville, Iowa). DNA oligos were ligated into the pENTR™/U6 using the BLOCK-iT™ U6 RNAi entry vector kit (Invitrogen, K4944-00) according to the manufacturer’s instructions to create the plasmid shRNAs. Plasmids were transformed in One Shot -TOP10 Chemically Competent E. coli cells (Invitrogen, C4040-10) according to the manufacturer’s directions. ShRNA Sequences were verified by DNA sequencing (AGCT, Inc, Wheeling, IL). NIH3T3 and HEK293 cells were purchased from ATCC, Inc, and maintained at 37°C in 5% (v/v) CO2 in DMEM/High glucose with 10% (v/v) FBS (Biowest, S01520) and 1x Antibiotic-antimycotic (100U/ml penicillin G, 100ug/ml streptomycin sulfate, 0.25ug/ml amphotericin B). Cells were passaged before reaching confluency. |
| siRNA and shRNA design |
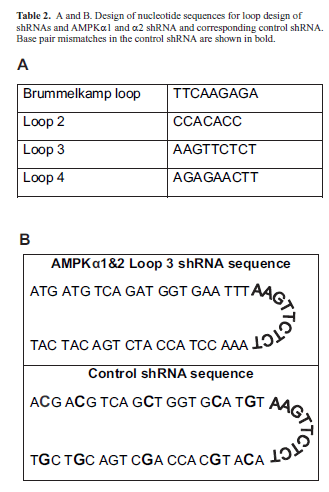
| cDNA sequences of human, mouse, and rat AMPKα1 and AMPKα2 were aligned in silico using the MacVector software program. The sequence ATGATGTCAGATGGTGAATTT was identified in the NCBI database (http:// www.ncbi.nlm.nih.gov/index.html) and determined to be 100% identical following alignment of all human, mouse, and rat cDNAs and corresponded to the following nucleotide regions: hAMPKα1 553-573, mAMPKα1 498-518, rAMPKα1 487-507, hAMPKα2 558-578, mAMPKα2 596-616, rAMPKα2 494-514. The global siRNA and shRNA were designed to target this region (Table 1A). A second sequence, AATGGAATATGTGTCTGGAGG, was 100% conserved in the cDNA sequences of human, mouse, and rat AMPKα2 (Table 1B). This region contains two base pair mismatches with the mouse and rat AMPKα1 sequences and three base pair mismatches with the human AMPKα1 sequence. A control shRNA was designed to be identical to the AMPKα1and 2 shRNA, with the exception of 5 nucleotide mismatches (Table 2B). The sequence of the control shRNA (ACGACGTCAGCTGGTGCATGT) did not contain significant homology to known genes in the human, mouse, or rat genomes as determined by analysis in the NCBI/BLAST program. shRNA stem loop design was based upon previous studies (Table 2A; Brown TL, unpublished data; Brummelkamp et al, 2002a). ShRNA oligonucleotides were annealed and cloned into the pENTR/U6 vector for propagation and use in experiments. |
| Transfection |
| For siRNA studies, cells were transfected with 100nM or 20nM siRNA with Lipofectamine 2000 according to the manufacturer’s instructions. For shRNA studies, HEK293 cells were seeded on 60mm plates at 1.2x106 cells/plate and transfected with a 3:1 ratio of Lipofectamine2000 (Invitrogen, 11668019) to DNA according to the manufacturer’s specifications. NIH3T3 cells were simultaneously seeded on 60mm plates at 8x105 cells/plate and transfected with a 5:1 ratio of Metafectene (Biontex, T020-1.0) to DNA according to the manufacturer’s specifications. Transfection efficiency was determined by replicate transfection with GFP and calculated as the relative number of GFP positive cells/total cell number. Cells were lysed 24, 48, or 72hrs post-siRNA transfection and 72hrs post-shRNA transfection for analysis. |
| Western blotting |
| Whole cell lysates were collected with Cell Lysis Buffer (Cell Signaling, 9803) supplemented with Complete Protease Inhibitor Tablets (Roche, #11836153001). Lysates were briefly sonicated on ice, and protein concentrations were determined by the Bradford method (Bradford, 1976; Gultice et al, 2009). Lysates were boiled in Laemmli reducing buffer and 50μg of total protein from each sample was electrophoresed on 10% (w/v) SDS polyacrylamide gels for AMPK isolation or 8% (w/v) SDS polyacrylamide gels for ACC isolation. Proteins were transferred to an Immobilon PVDF Membrane (Millipore, IPVH08100) and blocked in Tris-buffered saline containing 0.1% (w/v) Tween-20 and 5% (v/v) fat-free, dry milk. Antibodies purchased from Cell Signaling Technology (anti-AMPKα1 #2795; antitotal AMPKa #2603; anti-phosphoACC #3661; anti-total ACC #3676) were incubated overnight at 4°C at a dilution of 1:1,000 in Tris-buffered saline with 0.1% (v/v) Tween- 20 and 5% (v/v) bovine serum albumin. Antibodies purchased from GeneTex (anti-AMPKα2, GTX111373) were incubated overnight at 4°C at a dilution of 1:2,000 in Tris-buffered saline with 0.1% (v/v) Tween-20 and 5% (w/v) fat-free, dry milk. Anti-actin C4 monoclonal antibody (Seven Hills Bioreagents) was used at a 1/10,000 dilution for 1 hr at room temperature. Horseradish peroxidase-conjugated secondary antibodies were purchased from Promega (anti-rabbit IgG, HRP conjugate #W401B; and anti-mouse IgG, HRP-conjugate #W402B) and used at a dilution of 1:25,000 for 1hr at room temperature. Blots were developed using Super- Signal West Pico Chemiluminescent Substrate kit (Thermo Scientific, 34080), exposed to x-ray film, and visualized by chemiluminescence. |
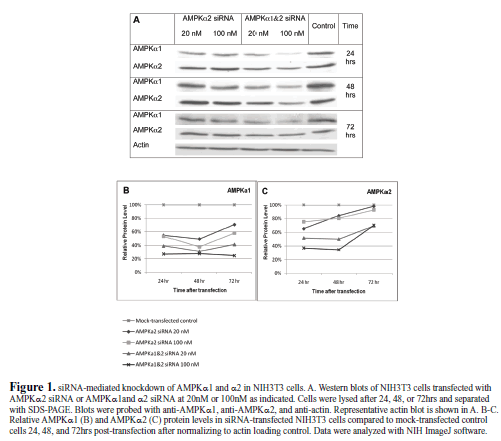 |
| Data analysis |
| Western blot data was analyzed using NIH Image J software (rsbweb.nih.gov/ij/) to quantitate protein levels. For all siRNA and shRNA experiments, a minimium of three independent experiments were performed. Statistical significance was determined using one-way ANOVA. |
 |
Results |
| siRNA-mediated knockdown of AMPKa |
| A number of siRNAs against AMPK have been generated (Neurath et al, 2006; Aguliar et al, 2007; Zhou et al, 2009; Vucicevic et al, 2009), but there is no reported siRNA that is able to knock down both catalytic isoforms of AMPKα1 and AMPKα2 in multiple species. To generate a global AMPKα1 and α2 gene knockdown, we aligned the cDNA sequences of human, mouse, and rat AMPKα1 and α2 and identified a single 21 bp sequence conserved in each (Table 1). We generated an siRNA targeting this sequence to determine if it could effectively knockdown AMPKα1 and AMPKα2 simultaneously. We also made an siRNA against a region in AMPKα2 that is specifically conserved in human, mouse, and rat (Table 1). The siRNAs were tested in mouse fibroblast cells, NIH3T3s, at two different concentrations (20nM and 100nM), and the cells were lysed 24, 48, and 72hrs post-transfection and run on a western blot (Figure 1). The siRNAs were tested at 100nM to determine general effectiveness and at 20nM for specificity (Semizarov et al, 2003). The global siRNA knocked down AMPKα1 and α2 protein levels at both concentrations at all three time points compared to mock-transfected control, particularly at 72 hrs post-transfection. The AMPKα2 siRNA caused a moderate knockdown of AMPKα1 and α2. Surprisingly, it caused a greater knockdown of AMPKα1 than AMPKα2, though the siRNA was not a perfect match for AMPKα1, although this may be a result of mismatch tolerance (Holen et al, 2002; Amarzguioui, 2003). These results prompted us to select the sequence of the global AMPKα1 and α2 siRNA to generate an shRNA that can be used to stably knockdown AMPKα1 and α2 for future studies. |
| shRNA-mediated knockdown of AMPKa |
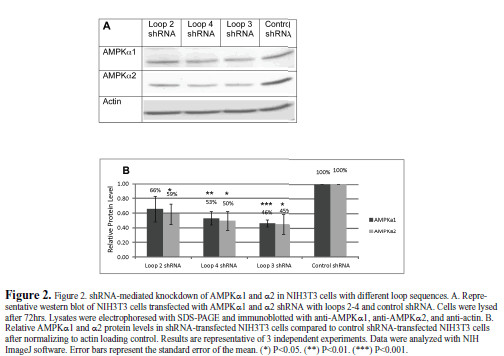
| Though the loop structure of an shRNA is cleaved prior to binding of the RNA to the target mRNA, the sequence of the loop can dramatically alter the activity of the shRNA (Schopman et al, 2010). We will refer to the most commonly used shRNA loop sequence (TTCAAGAGA) as the Brummelkamp loop (Brummelkamp et al, 2002b). However, this particular loop sequence begins with two thymidine residues. RNA polymerase III, which drives shRNA production from the U6 promoter in the pENTR vector, recognizes a stretch of four or more thymidine residues as a stop site (Geiduscheck et al, 1988). Therefore, this loop cannot be used if the stem sequence of the shRNA ends in two or more thymidine residues, as is the case for the AMPKα1 and α2 shRNA (see Table 1). For this reason, we needed to design alternative loop sequences that did not begin with thymidine (Table 2) that would effectively knockdown AMPK. We tested three loops, referred to as loops 2-4, in NIH3T3 cells (Figure 2). Two of the loops, loop 3 and loop 4, significantly knocked down protein levels of both isoforms of AMPKa. Loops 3 and 4 were the complement and the reverse, respectively, of the Brummelkamp loop. We found that the complement of the Brummelkamp loop, loop 3, resulted in the greatest knockdown, 54% knockdown of AMPKα1 and 55% knockdown of AMPKα2 and was thus chosen for further study. We created a control shRNA containing just five base-pair mismatches from the target 21nt siRNA sequence that also used the loop 3 sequence (Table 2). AMPKα1 and α2 protein levels, in cells transfected with the control shRNA, were equivalent to untreated, mock-transfected controls (data not shown). This control shRNA was included in all subsequent shRNA experiments. |
|
| Knockdown of AMPKa in mouse and human cells |
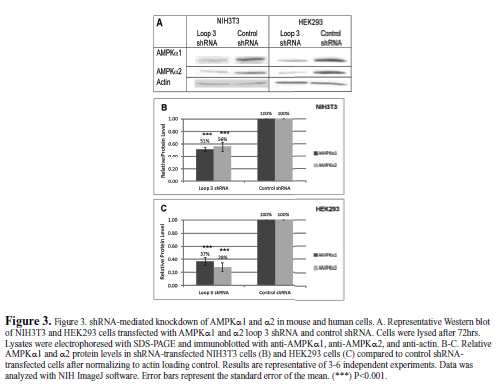
| We tested the shRNA in mouse NIH3T3 cells and human embryonic kidney HEK293 cells to demonstrate its ability to knock down AMPKα1 and α2 in different species (Figure 3). The loop 3 shRNA significantly knocked down protein levels of both isoforms of AMPKa in both species. In mouse NIH3T3 cells, AMPKα1 and AMPKα2 protein levels were knocked down 49% and 44%, respectively, compared to the control shRNA. In human HEK293 cells, AMPKα1 and AMPKα2 protein levels were knocked down 63% and 72%, respectively, compared to the control shRNA. The shRNA was likely more effective in human than mouse cells due to the increased transfection efficiency in HEK293 cells. |
| Functional knockdown of AMPKα |
| To demonstrate a functional knockdown of AMPK by the loop 3 global shRNA, we examined the phosphorylation of a direct AMPK target, ACC (acetyl-CoA carboxylase). Transfection of HEK293 cells with the loop 3 AMPKα1and α2 shRNA did not alter total ACC protein levels; however it did cause a significant reduction in the phosphorylated form of ACC (phosphoACC, Figure 4). The protein level of phosphorylated ACC was knocked down by 41% by the AMPKα1and α2 shRNA compared to the control shRNA.The total ACC protein levels in the AMPKα1 and α2 shRNA-treated cells compared to the control shRNAtreated cells were nearly identical and unaffected by transfection. |
 |
 |
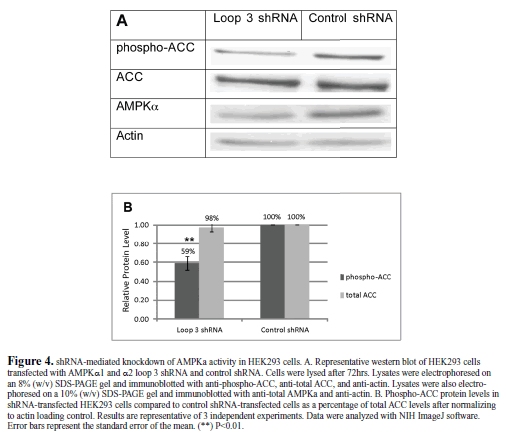 |
Discussion |
| AMPK is a key regulator of cellular metabolism, and it exerts its function through the catalytic alpha subunit. Knocking down both isoforms of the catalytic subunit is a way to study the function of AMPK; however, previous knockdowns of AMPKa were only effective in one model system (Neurath et al, 2006; Aguilar et al, 2007; Zhou et al, 2009; Vucicevic et al, 2009). This led us to develop an siRNA that can target both catalytic α1 and α2 isoforms of AMPK in human, mouse, and rat cells. Because siRNA only causes a transient knockdown in protein levels, we converted the siRNA into a plasmid-based shRNA. This shRNA caused a significant reduction in AMPKa protein levels in both human and mouse cells. We hypothesize the shRNA will also be very effective in rat cells because the sequence is 100% conserved. |
| When designing an shRNA, the loop that is used can have a significant impact on the knockdown of the target protein (Schopman et al, 2010). We chose to test three different loops to determine which is the most effective. The complement of the most commonly reported loop (loop 3) was found to be the most effective. This loop can be used for shRNAs that end in thymidine residues as an alternative to the Brummelkamp loop to prevent the formation of an RNA polymerase III stop site. |
| Knockdown of protein levels does not necessarily correspond to a reduction in activity of the target protein. For this reason, we wanted to ensure AMPKα activity was also reduced by the shRNA. ACC is phosphorylated at Ser79 by AMPKα (Ha et al, 1994). ACC catalyzes the production of malonyl-CoA from acetyl-CoA, thus providing the starting material for fatty acid synthesis. When ATP is depleted from the cell, ACC is inactivated by AMPK, and acetyl- CoA is used for energy production in the citric acid cycle. The reduction in phospho-ACC levels indicates that AMPK activity, not just protein levels, was reduced by the shRNA. |
| The shRNA used in this study was cloned into the pENTR/ U6 Entry plasmid-based shuttle vector. In the future, the shRNA from this plasmid can be transferred into other expression systems, such as the lentiviral Block-iT Dest vector (Invitrogen), which can then be used to make a lentivirus capable of producing the AMPK shRNA. This lentiviral delivery system also provides the ability for clonal selection using Blastocidin. Lentiviral infection with the shRNA should lead to higher levels and/or a greater number of cells with AMPK knockdown due to the dramatically higher efficacy that is achieved by viral transduction. |
Conclusions |
| • Identification of an efficacious single siRNA and subsequent shRNA to both alpha 1 and alpha 2 AMPK isoforms were identified in silico. |
| • Identification of alternate shRNA loop structures that facilitate significant knockdown |
| • Generation of an AMPK shRNA, that can significantly knockdown protein levels and activity of AMPKα1 and α2, was confirmed in cell culture |
| • Generation of an AMPK alpha 1 and alpha 2 shRNA, that can significantly knock down protein levels in multiple species, was confirmed in cell culture |
| • Further work will investigate in vivo efficacy in relevant animal models. |
Acknowledgements |
| This work was supported in part with grants from WSUCOSM (CNW and TLB), NIH-NHLBI R01#HL091836 to CNW and NIH-NICHD R01# HD059969 to TLB. Antiactin C4 antibody was kindly provided by Dr James Lessard from Cincinnati Children’s Hospital Medical Center. Thanks to Erica Baker, Renee Albers, and Rebecca Bricker for excellent technical assistance. |
Compteting Interests |
| None declared. |
Abbreviations |
| AMPK; 5′-adenosine monohposphate-activated protein kinase |
| PRKAA; protein-kinase, AMP-activated ACC; acetyl-CoA carboxylase |
References
|