- Journal of RNA and Genomics (2014) Research Article
Enzyme-triggered PEGylated siRNA-nanoparticles for controlled release of siRNA
| Peerada Yingyuadα, Mathieu Mévelα, Carla Prataα, Christos Kontogiorgisα,β, Maya Thanouα,β and Andrew D Millerα,β,* αImperial College Genetic Therapies Centre, Department of Chemistry, Imperial College London, London, SW7 2AZ, UK, βInstitute of Pharmaceutical Science, Franklin-Wilkins Building, 150 Stamford Street, King’s College London, London, SE1 9NH, UK |
| *Correspondence to: Andrew Miller, Email: a.miller07@btinternet.com, Tel: +44 787 963 5513 |
| Received: 11 December 2013; Accepted: 28 January 2014; Published: 28 January 2014 |
| © Copyright The Author(s). First Published by Library Publishing Media. This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/ by-nc/2.5). This license permits non-commercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Abstract
A key goal of our recent research efforts has been to develop novel ‘triggerable nanoparticle’ systems with real potential utility in vivo. These are designed to be stable from the point of administration until a target site of interest is reached, then triggered for the controlled release of therapeutic agent payload(s) at the target site by changes in local endogenous conditions or through the application of some exogenous stimulus. Here we describe investigations into the use of enzymes to trigger RNAi-mediated therapy through a process of enzymeassisted nanoparticle triggerability. Our approach is to use PEG2000-peptidyl lipids with peptidyl moieties sensitive to tumour-localized elastase or matrix metalloproteinase-2 digestion, and from these prepare putative enzyme-triggered PEGylated siRNA-nanoparticles. Our results provide initial proof of concept in vitro. From these data, we propose that this concept should be applicable for functional delivery of therapeutic nucleic acids to tumour cells in vivo, although the mechanism for enzyme-assisted nanoparticle triggerability remains to be fully characterized.
KEYWORDS |
| Liposomes, elastase, enzyme, triggerable nanoparticles, triggered release, peptide |
INTRODUCTION |
| A key goal in using nanoparticles is the functional delivery of small interfering RNA (siRNA) to target cells in vivo. Appropriate lipid-based nanoparticles typically require a “surface layer” of polyethylene glycol [PEG]) that provides for colloidal stability in biological fluids and resistance to immune system challenge. However, the functional delivery of siRNA can be substantially affected by the presence of the surface PEG layer (Kostarelos and Miller, 2005; Miller, 2008a; Miller, 2008b). Therefore, we have become very interested in developing nanoparticles that are triggerable (i.e., stable in biological fluid, but triggered for the controlled release of therapeutic agent payload(s) at the target sites of interest by changes in local endogenous conditions or through the application of some exogenous stimulus). Previously, we reported pH-triggered PEGylated siRNA nanoparticles (known as pH-triggered siRNA-ABC nanoparticles; where A is entrapped siRNA, B represents lipid components, C is PEG surface layer) (Carmona et al, 2009). Thereafter, we recently described both a lipid-based and a polymer-based nanoparticle system, respectively, for the functional delivery of plasmid DNA (pDNA) to murine lung in vivo. The first system was categorized as a half-life-triggered nanoparticle system and the second as a redox-triggered nanoparticle system (Drake et al, 2010; Aissaoui et al, 2011). Here, we describe our efforts to devise lipid-based, enzyme-triggered siRNA-nanoparticles for enhanced functional delivery of siRNA, informed by the previously reported experiences of Pak et al and Hatakeyama et al in the use of enzymetriggering for functional delivery of active pharmaceutical ingredients (Pak et al, 1998; Pak et al, 1999; Hatakeyama et al, 2007). |
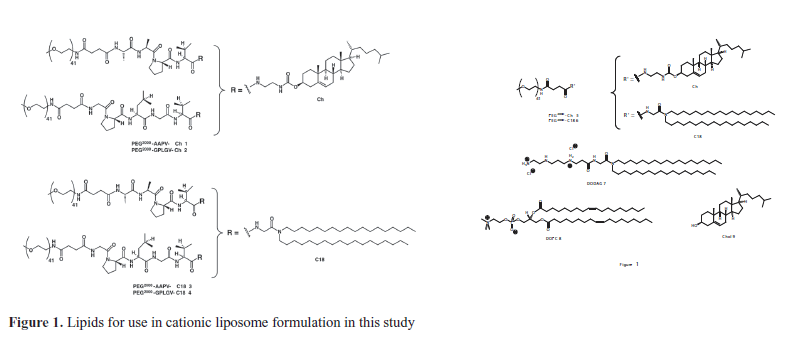
| As with previous reported experiences, our approach was to take advantage of the binding and cleavage specificity of tissue-matrix associated enzymes, such as human leukocyte elastase (HLE) and matrix metalloproteinase-2 (MMP-2), that are present in the extracellular spaces of tumour volumes. High levels of the proteolytic enzyme, elastase, are found in tumours in order to promote invasion and metastasis by degrading basement membrane and extracellular matrix barrier (Pak et al, 1998; Pak et al, 1999; Hatakeyama et al, 2007). On the other hand, many tumours are also well known to secrete substantial quantities of MMP2 for the degradation of the intercellular collagen matrix in order to promote invasion and metastasis (Hyuga et al, 1994; Lin et al, 2000; Morgunova et al, 2002; Wang et al, 2005; Lu et al, 2008; Kean et al, 2009; Han and Zhu, 2010; Stellas et al, 2010). Based upon proteolytic, amino acid residue consensus sequences of these two enzymes, we synthesised four different PEG2000-peptidyl-lipids 1-4 and two PEG2000-lipid controls 5 and 6 (Figure 1) that were used to prepare PEGylated siRNA-nanoparticles. We now report on a sequence of nanoparticle characterization studies and studies involving functional siRNA delivery to two cell lines in vitro. The results provide a clear demonstration for nanoparticle-mediated enzyme-triggered functional delivery of siRNA into cells, results that sit well with recent proof of concept data that were obtained for nanoparticle mediated enzyme-triggered functional delivery of pDNA to cells using comparable PEGylated pDNA-nanoparticles (Yingyuad et al, 2013). |
MATERIALS AND METHODS |
| General Procedure |
| Full details for the syntheses of PEG2000-peptidyl-lipids 1-4 and two PEG2000-lipid controls 5 and 6 are described elsewhere (Yingyuad et al, 2013). The cationic lipid N’,N’- dioctadecyl-N-4,8-diaza-10-aminodecanoyl-glycylamide (DODAG) 7 was prepared as described previously (Mevel et al, 2010). Dioleoyl-L-α-phosphatidylcholine (DOPC) 8 and cholesterol (Chol) 9 along with all other chemicals were purchased from Sigma-Aldrich, Lancaster or Merck Biosciences. Anti-luciferase siRNA was obtained from Qiagen. Silencer negative control siRNA was purchased from Applied Biosystems/Ambion. HLE was purchase from Sigma/Aldrich (UK), MMP-2 from Calbiochem (UK). Propidium iodide and SYBR® green II RNA gel stain were obtained from Molecular Probes (UK). SYBR® safe DNA gel stain and 10x Tris-borate-EDTA (TBE) electrophoresis buffer were purchased from Invitrogen. |
| Preparation of siRNA |
| Synthetic siRNA was dispersed in nuclease-free water to give a stock siRNA concentration of 50mM then stored at -20oC before use. |
| Preparation of siRNA lipoplex nanoparticles and PEGylated siRNA nanoparticles |
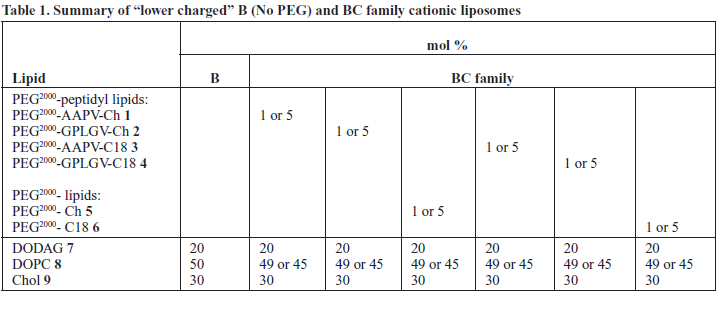
| PEG2000 lipids 1-6, DODAG 7, DOPC 8, Chol 9 and were prepared as stock solutions in CHCl3 and stored at -20oC. Appropriate volumes of each lipid stock were combined in a round bottom flask (5ml) containing CHCl3 (500ml). The solvent was slowly removed in vacuo to form an even lipid film that was then purged with N2 (g) to remove residual traces of organic solvent. The film was re-hydrated with 4mM 2-[4-(2-hydroxyethyl)-piperazin-1-yl]-ethanesulfonic acid (HEPES) (pH 7.0) to obtain a total lipid concentration of 1mg/ml. Lipid suspensions were subsequently subjected to sonication at 40oC for 40min, leading to the formation of uniform, unilamellar PEGylated cationic liposomes of the BC family (see Table 1). PEGylated siRNA nanoparticles were then prepared by mixing the appropriate volume of siRNA stock (50μM) with the resulting liposome solutions under heavy vortex mixing conditions to obtain siRNA nanoparticles at the desired lipid/siRNA ratios given in terms of a charge ratio that is calculated as 1.7x ([DODAG]/ [nucleotide]) (final [siRNA] typically 100μg/ml, 7μM). Corresponding siRNA-lipoplex nanoparticles were prepared in a similar way combining unilamellar cationic liposomes (B No PEG) instead (see Table 1) with the same siRNA stocks (50μM) as above. |
| Determination of siRNA entrapment efficiencies and nanoparticle properties |
| Propidium Iodide (PI) assay |
| PEGylated siRNA nanoparticles and appropriate corresponding siRNA lipoplex nanoparticles were prepared, as above, by combining various added volumes of cationic liposome suspensions (1mg/ml) with fixed siRNA aliquots (50pmoles in 6.6μl, 7μM). Resulting lipid/siRNA charge ratios were 0.5:1, 1:1, 2:1, 4:1 or 8:1. After 10min incubation at ambient temperature, all nanoparticles were diluted in 4mM HEPES buffer, pH 7.0 (total final volume 100μl, final [siRNA] 0.45μM). PI solution (100μl, 2.5μM) was then added and the mixtures were incubated at 37oC for 5min. The fluorescence intensity (Aex 535nm, Iem 617nm) of each mixture was measured using a Varioskan flash microplate reader. The fluorescence intensities of corresponding samples of BC PEGylated liposomes, or simple B (No PEG) cationic liposomes as appropriate, alone in PI solution (with no siRNA present) and of PI solution alone were also subtracted from fluorescence intensity data as background. The percentage of siRNA entrapped (%en-siRNA) was then determined using the following expression (1): |
 |
 |
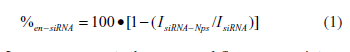 |
| where IsiRNA-NPs represents the measured fluorescence intensity of a given solution of nanoparticle complexed siRNA incubated with PI solution, and IsiRNA the measured fluorescence intensity of a corresponding concentration matched control solution of free siRNA also incubated with PI solution. |
| Agarose gel electrophoresis |
| PEGylated siRNA nanoparticles and appropriate corresponding siRNA lipoplex nanoparticles were prepared as for the PI assay using lipid/siRNA charge ratios of 0.5:1, 1:1, 2:1, 4:1 or 8:1. After 10min incubation at ambient temperature, the nanoparticles were diluted in 4mM HEPES, pH 7.0 (total final volume 100μl, final [siRNA] 0.45μM). SYBR® green II RNA gel stain (5μl of 10000x) was added and then aliquots of nanoparticle solutions (60ng in 10μl, 4pmol siRNA/well) mixed with 6x orange DNA loading dye (1μl) were loaded into each well of 0.8% (w/v) agarose gels. Free siRNA was used as siRNA marker. Electrophoresis was performed at 65mV for 30min and the gel was visualized under UV light. |
| Size and zeta potential measurements |
| Sizes of all cationic liposomes or siRNA lipoplex nanoparticles and PEGylated siRNA nanoparticles were measured by dynamic light scattering using a Delta N4+440SX particle analyzer (Coulter). Scattering was detected at 25oC using a 90o scattering angle. Mean nanoparticle diameters were determined by calculation from unimodal size distributions. The zeta potential measurements were performed on a Nanoseries Nano-ZS zetasizer (Malvern, UK) equipped with a 4mW He-Ne laser (633nm) and avalanche photodiode detector. All samples were prepared in 4 mM HEPES buffer, pH 7 (total lipid concentration used was 0.5mg/ml). All siRNA nanoparticles studied were prepared with a lipid/ siRNA charge ratio of 4 for this particular study. |
| siRNA nanoparticle-mediated gene knockdown efficiencies |
| Exogeneous gene expression |
| MCF-7 and HT1080 cells were seeded respectively at 3.5 × 104 per well for 72hr and at 2.5 × 104 cells per well for 24hr in 48-well plates (250ml of complete media) prior to transfection. The cells were grown at 37oC in a humidified, 5% CO2 (v/v) incubator until 80% confluent (MCF-7) or until 60% confluent (HT1080). Media was then removed and replaced with fresh media. Transfection with pEGFPLuc DNA was mediated by jetPEI™ (PolyPLUS) according to the manufacturer’s protocol. Briefly, pDNA (0.5mg) and jetPEI™ (1ml) were diluted separately in NaCl buffer (150mM, 25ml). An appropriate aliquot of jetPEI™ solution was added to pDNA solution, vortex mixed, and incubated at ambient temperature for 30 min. The mixture (50ml) was then added to the wells and MCF-7 or HT1080 cells, as selected, were incubated at 37oC in 5% CO2 (v/v) atmosphere for 2hr. The media was removed and the cells were washed with phosphate-buffered saline (PBS) (2 × 250ml) before being used for siRNA knockdown experiment. |
| Transient gene knockdown |
| pEGFPLuc transfected MCF-7 cells were seeded in 48-well plates (3.5 × 104 cells per well, 250ml of complete growth media) for 72hr prior to delivery of siRNA. The cells were grown until 80% confluent at 37oC in 5% CO2 (v/v) atmosphere. PEGylated siRNA nanoparticles were formulated from PEG2000-AAPV-Ch 1, PEG2000-AAPV-C18 3, PEG2000- Ch 5 or PEG2000-C18 6 as appropriate. Corresponding siRNA lipoplex nanoparticles were also prepared for control comparisons. All siRNA nanoparticles were prepared with a lipid/siRNA charge ratio 4. HLE (10ml, 1.03mM) was added to one complete set of nanoparticles and incubated at room temperature for 10min, while a second set contained no HLE. These mixtures were added to different wells containing pEGFPLuc transfected MCF-7 cell lines (250ml of complete media; final [siRNA] 15pmol/well) and the plates were then incubated at 37oC in 5% CO2 (v/v) atmosphere for 6hr. The media was then removed, the cells were washed with PBS (2 × 250ml) and the fresh media was added (250ml). The cells were incubated for a further 36hr before kit analysis (Promega, USA) of luciferase activity (expressed in relative light units [RLU, measured on Berthold Lumat LB 9507 luminometer] per mg of total protein content [determined by bicinchoninic acid assay, Pierce Thermo Scientific]). Relative extents of luciferase protein knock down levels were expressed as a percentage (%Luc-KD) according to expression (2): |
| where nantiLuc-siRNA is the luciferase activity measured post anti-luciferase siRNA delivery and ncontrol-siRNA the luciferase activity measured post control siRNA delivery. |
| In a second experiment of this type, pEGFPLuc transfected HT1080 cells were seeded in 48-well plates (2.5 × 104 cells per well, 250ml of complete media) for 24hr prior to delivery of siRNA. The cells were grown until 60% confluent at 37oC in 5% CO2 (v/v) atmosphere. In the meantime, PEGylated siRNA nanoparticles were formulated from PEG2000-GPLGV-Ch 2, PEG2000- GPLGV -C18 4, PEG2000- Ch 5 or PEG2000-C18 6 as appropriate. Once again, corresponding siRNA-lipoplex nanoparticles were also prepared for control comparisons. All siRNA nanoparticles were prepared with a lipid/siRNA charge ratio 4. Thereafter mixtures were added to different wells containing the pEGFPLuc transfected HT1080 cell lines (250ml of complete media; final [siRNA] 15pmol/well) and the plates were then incubated at 37oC in 5% CO2 (v/v) atmosphere for 24hr. Thereafter, media was removed, the cells were washed with PBS (2x 250ml) and fresh media was added (250ml). The cells were incubated for a further 36hr before analysis for luciferase activity. Relative extents of luciferase protein knock down knock down levels were determined in the same way as above. |
| Endogenous gene expression |
| MCF-7 and HT1080 cells were seeded at 1.2 × 105 and 8 × 104 in 6-well plates (2ml of complete growth media) for 72 and 24hr prior to transfection, respectively. The cells were grown until 80% confluent for MCF-7 and 60% confluent for HT1080, at 37oC in 5% CO2 (v/v) atmosphere. The media was removed and replaced with fresh media. The transfection of pUbC-Luc-S/MAR DNA was carried out using jetPEI™. Briefly, pDNA (3 mg) and jetPEI™ (6ml) were diluted separately in NaCl buffer (150 mM, 100 mL). A jetPEI™ solution aliquot was added to pDNA solution, vortex mixed, and incubated at room temperature for 30min. The mixture (200 ml) was added to the wells and incubated at 37oC in 5% CO2 (v/v) atmosphere for 2hr. The media was removed, washed with PBS (2 × 2 ml) and the fresh media was added (2ml). The cells were incubated for a further 48hr. The media was removed and washed with with PBS (2 × 2 ml). The fresh media containing G418, a selection drug, (1mg/ml) was added (2ml) to each well and the plate was maintained at 37oC in 5% CO2 (v/v) atmosphere. The antibiotic-containing media should be replaced every 3-4 days. Drug-resistant and endogeneous luciferase expressing colonies of MCF-7 (MCF-7-luc) appeared in 4 weeks. Drugresistant and endogeneous luciferase expressing colonies of HT1080 (HT1080-luc) appeared in 2 weeks after transfection. In each case media was removed and cells washed with PBS (2 × 2 ml). Trypsin-EDTA (300ml) was added to detach the cells and individual MCF-7-luc or HT1080-luc colonies were transferred into a new 6-well plate containing complete media. These cells were incubated at 37oC in 5% CO2 (v/v) atmosphere and media replaced every 3-4 days. When confluent, MCF-7-luc or HT1080-luc cells were trypsinized and transferred to a 75ml tissue culture flask containing complete media. The cell lines were analysed for luciferase expression before entering siRNA-mediated knockdown experiments as above. |
| Endogenous gene knockdown |
| This was carried out in an equivalent way to transient gene knockdown except that experiments were performed with MCF-7-luc or HT1080-luc cells instead. |
RESULTS |
| Preparation of siRNA nanoparticles |
| Samples of siRNA (A component) were formulated as indicated with cationic liposomes (B component, no PEG) to give siRNA-lipoplex nanoparticles (known here as siRNAAB nanoparticles). In addition, samples of siRNA (A component) were also formulated with PEGylated BC family cationic liposomes to yield PEGylated siRNA nanoparticles (known here as siRNA-ABC nanoparticles). All cationic liposomes or PEGylated cationic liposomes were prepared throughout using the cationic lipid N’,N’-dioctadecyl- N-4,8-diaza-10-aminodecanoylglycylamide (DODAG) 7 (Mevel et al, 2010). |
| Physical Properties of siRNA nanoparticles |
| The process of siRNA encapsulation was followed at different lipid/siRNA ratios. In comparison to pDNA constructs that are approximately 4000bp at a minimum (Bloomfield, 1991), siRNA is much smaller consisting of 19-21 base pair (bp) duplexes. Therefore siRNA has many far fewer negative charges – a maximum of 42 negative charges per molecule – while pDNA has approximately 8000 negative charges. In consequence, whereas lipid-pDNA formulations are often reported in terms of lipid/pDNA w/w ratios, we report lipid/siRNA ratios in the form of an N/P charge ratio where N corresponds with net cationic charges presented by lipid formulations (1.7 per molecule of DODAG) and P the number of anionic charges presented by siRNA (1 per nucleotide; 2 per bp). |
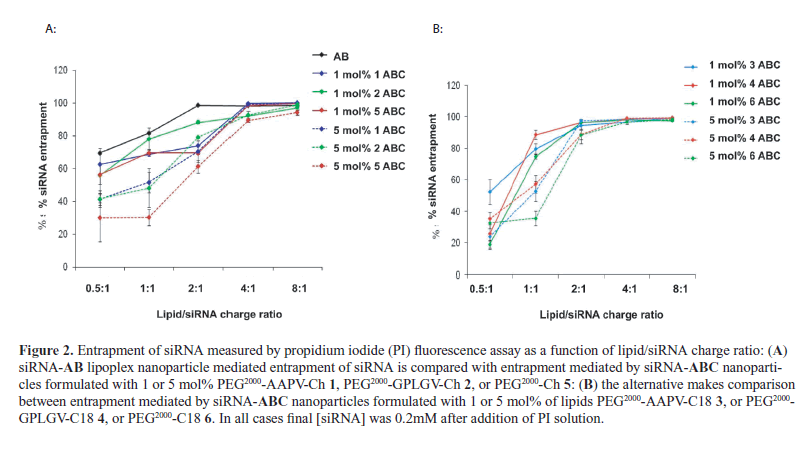
| The efficiency of siRNA entrapment in combination with simple cationic liposomes (B, no PEG) or PEGylated cationic liposomes (BC family) (Table 1) appeared quite variable at low lipid-siRNA charge ratios but converged agreeably at charge ratios of 4 and above (Figures 2a and 2b). Similar findings were revealed by gel retardation assay as well, although complete retardations were only observed at charge ratios of 8 (Figure 3). Arguably, at ratios |
 |
 |
| 4 some entrapped siRNA may be surface associated, then fully entrapped at ratios of 8 and above. Indeed, such an argument would be consistent with observations of Buyens et al, who characterized some surface binding of siRNA to PEGylated cationic liposomes following classical mixing of both components (Buyens et al, 2009). |
| Confirmed size and z-potential measurements also suggested that siRNA encapsulation by BC family PEGylated cationic liposomes can result in siRNA-ABC nanoparticles that are both smaller in dimensions, lower in polydispersity and lower in charge than corresponding pDNA-nanoparticles (Figure 4) (Yingyuad et al, 2013). Note in particular that nanoparticles formulated with 5mol% PEG2000 possessed diameters <120nm in size and z-potentials that were almost zero, in line with requirements for in vivo applications involving cancer (Kamaly et al, 2008; Kamaly et al, 2009; Kamaly et al, 2010; Kenny et al, 2011). In other experiments (reported elsewhere) with pDNA delivery we observed that equivalent 1-5mol% PEGylated pDNA-ABC nanoparticles were stable with respect to colloidal instability and aggregation when incuof |
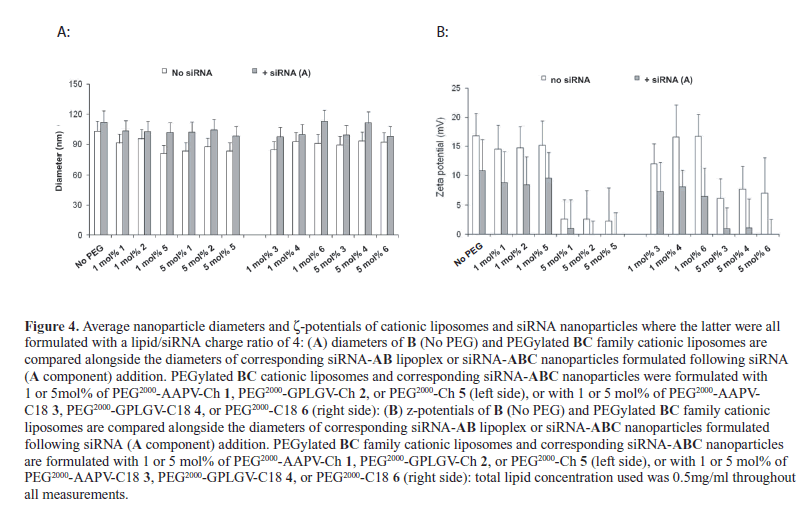 |
| bated at 37oC for 4hr in the presence of 10% (v/v) fetal calf serum (FCS). Moreover, 5mol% PEGylated pDNAABC nanoparticles were found stable even if incubated at 37oC for 4hr in the presence of 80% (v/v) FCS (Yingyuad et al, 2013). Therefore, our 1-5mol% PEGylated siRNAABC nanoparticles reported here are expected to possess equivalent stability profiles. |
| SiFection Properties of siRNA-nanoparticles |
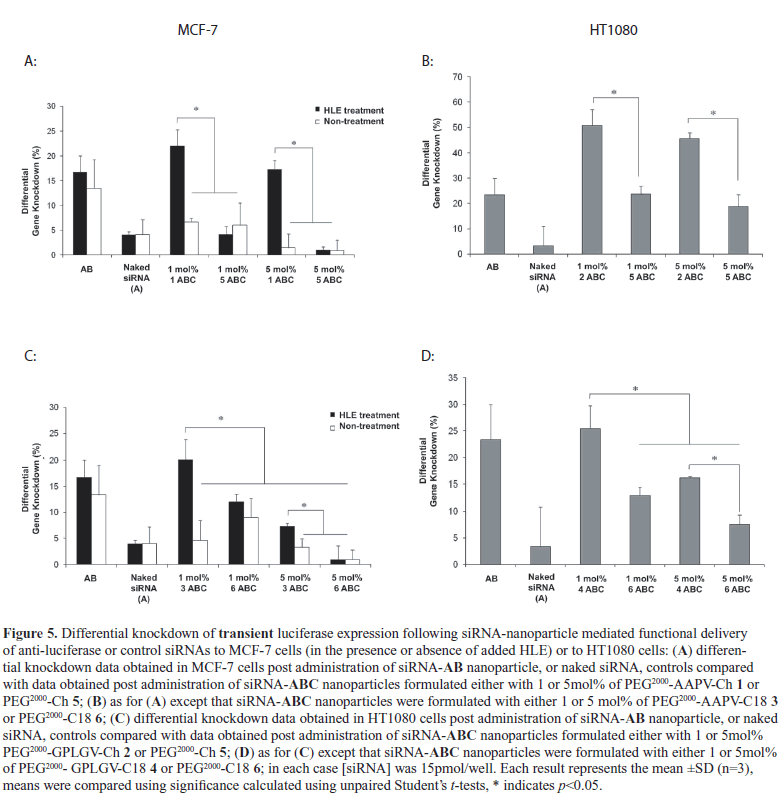
| Having prepared discrete siRNA-ABC nanoparticles, the next stage was to obtain proof of concept data for functional siRNA-mediated exogeneous and endogenous gene knockdown in vitro, under conditions that might at least simulate in vivo conditions in the vicinity of tumour target cells. These studies were performed with MCF-7 cells (breast cancer cell line, expressing HLE protein) and HT1080 cells (primate fibrosarcoma cell line, expressing MMP-2 protein). Given the inconsistent expression of HLE by MCF-7 cells, functional siRNA delivery experiments were always peformed with and without added exogenous HLE. HT1080 cells were shown comfortably to secrete MMP-2 (7ng/ ml of culture medium; MMP-2 human ELISA assay kit, Invitrogen). |
| Functional delivery of siRNA (siFection) was studied by means of luciferase knock-down experiments that were set up to monitor specific RNAi versus control RNAi mediated effects in cells of interest looking in particular for differential effects on enzyme levels (Figures 5 and 6). All siRNA-AB lipoplex nanoparticles and siRNA-ABC nanoparticles tested were formulated at a preferred lipid/ siRNA charge ratio of 4 for functional delivery of either specific anti-luciferase or a non-specific control siRNA. In all cases, the differential knockdown effects were always enhanced when siRNA-ABC nanoparticles were prepared from PEG2000-peptidyl lipids in preference to corresponding control PEG2000-lipids. Critically, all siRNA functional delivery experiments reported here were performed in complete medium with added 10% (v/v) fetal calf serum (FCS) so that cell line expressed enzymes should be secreted functional. These simulated in vivo-conditions are typically too severe for routine synthetic-nanoparticle mediated functional delivery of nucleic acids in vitro (Keller et al, 2003; Mevel et al, 2010; Yingyuad et al, 2013). |
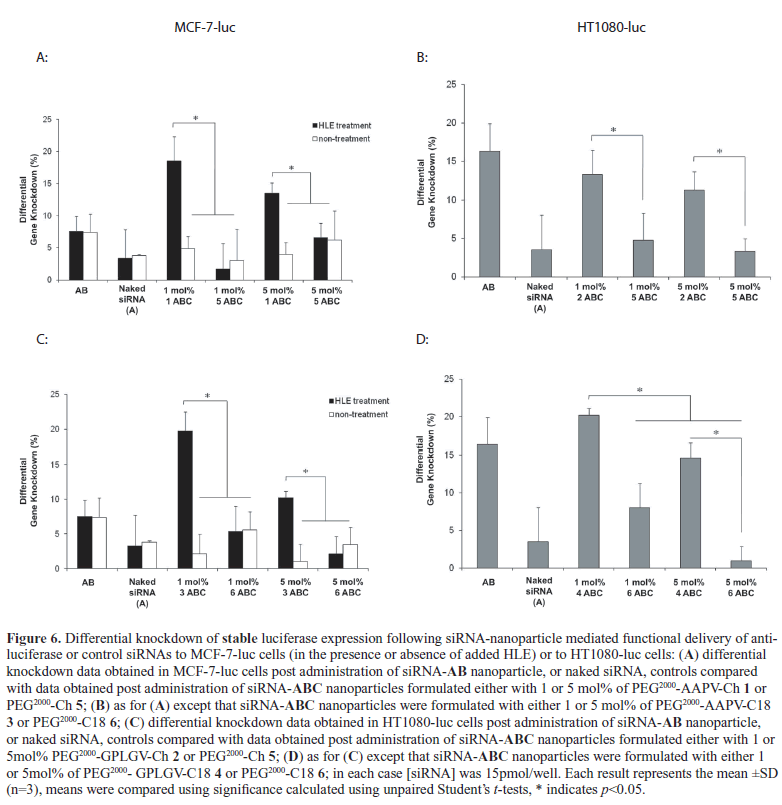
| Nevertheless, differential siRNA-mediated knockdown effects were observed with all siRNA-ABC nanoparticles studied, most especially when nanoparticles were prepared with PEG2000-peptidyl lipids as opposed to control PEG2000-lipids. Those siRNA-ABC nanoparticles prepared with control PEG2000-lipids were observed to promote levels of differential siRNA-mediated knockdown 4-10 fold lower than corresponding siRNA-ABC nanoparticles prepared with PEG2000-peptidyl lipids. Most striking, 5mol% siRNA-ABC nanoparticles prepared with PEG2000-peptidyl lipids were shown capable of mediating differential siRNAmediated knowndown of endogenous/stable luciferase expression to extents equivalent with if not better than knockdown effects observed from administration of corresponding positive control siRNA-AB lipoplex nanoparticles (Figure 6). Given that 5 mol% siRNA-ABC nanoparticles should be the most compatible for in vivo RNAi effector delivery according to our previous data analysis (Carmona et al, 2009; Kenny et al, 2011) and the foregoing analysis above, then these data provide proof of concept that 5 mol% siRNA-ABC nanoparticles formulated with PEG2000-peptidyl lipids should be capable of mediating functional delivery of siRNA to tumour tissues in vivo as well. |
 |
DISCUSSION |
| A critical objective of the work described here was to demonstrate that enzyme-triggered pDNA-ABC nanoparticles (Yingyuad et al, 2013), could be reformulated as siRNAABC nanoparticles suitable for the enzyme-triggered functional delivery of siRNA to cells instead of pDNA. Indeed, all our data (Figures 2-6) suggest that this main objective was achieved. In particular, 5mol% siRNA-ABC nanoparticles prepared with 5mol% PEG2000-peptidyl lipids 1, 2, 3 or 4, were shown to mediate gene knockdown as efficiently, if not more efficiently than siRNA-AB nanoparticle (siRNAlipoplex nanoparticle) positive controls, irrespective of whether the gene target concerned was an exogenously expressed transgene or a model endogenous gene (Figures 5 and 6). The importance of these observations is that 5mol% siRNA-ABC nanoparticles should also be appropriate for in vivo use given the similarity of their physical properties and lipid compositions to other pH-triggered and theranostic 5 mol% siRNA-ABC nanoparticles that have been shown capable of functional siRNA delivery in vivo (Kenny et al, 2011; Kolli et al, 2013). |
| The exact mechanism of the enzyme-trigger remains to be determined. There remain two main alternative possibilities as follows: |
| 1. Enzyme-assisted cleavage of peptide linkages could take place thereby liberating PEG moieties from nanoparticle surfaces but leaving sufficient PEG to prevent colloidal instability (Terada et al, 2006). Such partial removal of PEG would then lead to the exposure of peptide moieties at nanoparticle surfaces with N-terminal positive charges that could promote cellular internalization. Indeed, nanoparticle surface exposed peptides and proteins themselves are already known to promote nonspecific enhanced cell uptake for reasons that remain to be fully established (Waterhouse et al, 2005). |
 |
| 2. The peptide sequences themselves could act as temporary sequestration/binding sites for enzymes that over time could hydrolyze the linkage leading to PEG loss, but in the shorter term allow proteins to coat the nanoparticle surface and promote transfection efficiency in a similar way to that recounted above. |
| In partial support of the first possibility, when PEG2000- AAPV-Ch 1 (0.4mM) was incubated in neutral buffer (10mM HEPES, 154mM NaCl, 0.1mM EDTA, pH 7.4) at 37oC, in the presence of HLE (0.05mM), digestion products were observed by HPLC using an evaporative light scattering detector (ELSD). However, this digestion process took place only slowly and inconsistently over a 2hr period (data not shown). Similarly when digestion studies were carried out with MMP-2 and PEG2000-GPLGV-Ch 2 in buffer (as above but without EDTA) at pH 8.0, similar mixed results were obtained once again (data not shown). Potentially serum enzymes may also be able to affect some non-specific cleavage of peptide linkages to augment enzyme-specific cleavage |
| Overall, our siRNA-mediated gene knockdown data also suggest that there is little to choose between PEG2000- peptidyl lipids prepared from N1-cholesteryloxycarbonyl- 1,2-diamine (Ch) or N, N-dioctadecylglycylamide (C18) lipid moities. Since cholesterol based PEG lipids are likely to have a much more limited nanoparticle residence times than N, N-dioctadecylglycylamide based PEG lipids (Aissaoui et al, 2011), then the former series might be expected to dissociate more easily from siRNA-ABC nanoparticles hence providing an additional form of assistance to functional siRNA delivery. However, the active contribution of such a putative mechanism seems minimal at best. |
| Irrespective of mechanism, further development of enzymetriggered delivery of siRNA could involve increasing the sensitivity of peptide linkers to create more efficient PEG release or surface binding of proteins which would ultimately increase siRNA-mediated gene knockdowns. For instance, the numbers of amino acid residues involved could be increased to improve interaction with enzyme active sites. Furthermore, peptide sequences including GPLGIAGQ (Terada et al, 2006) and GPLGVRGC (Harris et al, 2006) could be employed that were demonstrated recently to be efficient MMP-2 target sequences. The use of such octapeptides may not only allow for more efficient removal of PEG via enzymatic cleavage of the peptide linker but also enhance intracellular trafficking by promoting internal endosomolysis for example. Thereafter, the inclusion of bona fide biological targeting ligands attached to the surfaces of PEGylated nanoparticles could provide a further boost to functional delivery at target cells of interest (Cheon et al, 2009; Kamaly et al, 2009). |
| Finally, one other noteworthy aspect of our studies reported here has been the use of MCF-7-luc and HT1080-luc cells selected for stable luciferase expression. These cell lines were obtained by means of transfection using pDNA comprising luciferase reporter gene, a G418 antibiotic resistant gene and an S/MAR sequence. Inclusion of S/MAR sequences into pDNA constructs has been shown previously to confer sustained epichromosomal transgene expression in both in vitro and in vivo (Argyros et al, 2008). Post transfection with G418-containing pDNA, cell-lines were kept under antibiotic selection over a period of two weeks in order to promote the establishment of stable luciferase expression as if from an endogeneous gene (level of luciferase expression was found to be ~108RLU/mg protein). Stably expressing cells lines were found initially to enable facile differential siRNA-mediated endogenous gene knockdown studies with the luciferase gene as target. Another clear advantage is that these same cell-lines allow for the generation of simpler, cost-effective, animal models of cancer comprising xenograft tumours that stably express luciferase as well. Once such tumours are established, functional delivery of siRNA to tumour cells can be studied with ease in vivo by comparing tumour bioluminescence levels before and after nanoparticle- mediated delivery of anti-luciferase or control siRNAs to tumour cells, by analogy with the use of biolumnescence in our recent liver work (Argyros et al, 2008; Kolli et al, 2013). |
CONCLUSION |
| Overall, we formulated PEGylated siRNA-nanoparticles that appear to possess enzyme triggerability, namely they are primed for enzyme-triggered, functional siRNA delivery. However, the exact mechanism of this enzyme-triggered process is yet to be firmly established. Two alternative possible mechanisms have been proposed. Either proposed mechanism is consistent with the definition of nucleic acid-nanoparticle triggerability, namely that a nanoparticle should be stable until a chosen target site is reached and then triggered once there for controlled release of entrapped therapeutic nucleic acids by an endogenous or exogeneous trigger. |
ACKNOWLEDGEMENTS |
| We thank the Mitsubishi Chemical Corporation and IC-Vec Ltd for the provision of financial support for the Imperial College Genetic Therapies Centre. P.Y. wishes to thank the Royal Thai Government for a Ph.D. studentship. |
COMPETING INTERESTS |
| None declared. |
LIST OF ABBREVIATIONS |
| siRNA; small interfering RNA |
| PEG; polyethyleneglycol |
| HLE; human leukocyte elastase |
| MMP-2; matrix metalloproteinase-2 |
| DODAG; N’,N’-dioctadecyl-N-4,8-diaza-10-aminodecanoylglycylamide |
| DOPC; dioleoyl-L-a-phosphatidylcholine |
| Chol; cholesterol |
| HEPES; 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesu ethanesulfonic acid |
| PI; propidium iodide |
| FCS; fetal calf serum |
| DMEM; Dulbecco’s Modified Eagle’s Medium |
| PBS; phosphate-buffered saline |
| DOPE; dioleoyl-L-a-phosphatidylethanolamine |
| EDTA; ethylenediamine tetraacetic acid |
References |
|