- Journal of RNA and Genomics (2010) Technology Report
A streamlined sub-cloning procedure to transfer shRNA from a pSM2 vector to a pGIPZ lentiviral vector
| Sara Ansaloni, Nadav Lelkes, Jonathan Snyder, Charles Epstein, Aditi Dubey and Aleister J Saunders* Department of Biology, Drexel University, Philadelphia, PA, USA |
| *Correspondence to: Aleister Saunders, Email: Aleister.Saunders@Drexel.edu, Tel: +1 215 895 6772, Fax: +1 215 895 1273 |
| Received 26 August 2010; Revised 12 October 2010; Accepted 13 October 2010; Published online 27 October 2010 |
| J RNAi Gene Silencing (2010), 6(2), 411-415 |
| © Copyright The Authors: This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/). This license permits noncommercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Abstract
RNA interference (RNAi) is a widely used molecular biology technique to investigate the importance of specific genes in molecular pathways. Since mammalian cells are equipped with endogenous RNAi processing machinery, it has become common practice to transfect constructs that encode for short hairpin RNAs that are then cleaved to form the active RNAi sequences that bind to target mRNAs. Given the profit potential of this research approach, companies have developed retroviral libraries of shRNA constructs targeting the majority of the human genes. Recent technologic advances have allowed the rapid improvement of the vectors carrying the shRNA constructs while the silencing sequences remain the same. Therefore, subcloning of shRNA sequences from more obsolete vectors to newer vectors is a straightforward way to take advantage of newer delivery technologies. We describe here a streamlined procedure to transfer shRNA sequences from the pSM2 retroviral vector to a newer pGIPZ vector that is more stable, contains a GFP cassette and allows the preparation of high titer viral particles for transduction of cells and in vivo use. We demonstrate that our protocol provides a cost-effective and fast method to successfully sub-clone shRNA from a pSM2 retroviral vector to a pGIPZ lentiviral vector making it a useful tool for the investigators that have purchased pSM2 vectors in the past and wish now to upgrade their constructs by inserting them in more versatile vectors.
Keywords |
| Sub-cloning, shRNA, RNA interference, lentivirus, retrovirus, pSM2, pGIPZ, pTRIPZ |
Introduction |
| RNA interference (RNAi) is an endogenous system that regulates gene expression. Since its discovery (Fire et al, 1998) it has been exploited to silence specific genes and has become an important experimental method utilized in cellular and in vivo studies. RNAi based therapies are in development (Castanotto and Rossi, 2009). Given this promise and utility, many current investigations aim at better understanding the molecular mechanisms of RNAi and to find effective delivery methods for RNAi reagents. |
| There are several approaches to introduce silencing RNAs into cells. One of them is to directly introduce short interfering RNAs, 21-23 nucleotide duplexes targeting specific mRNAs, into the cells or tissue under investigation (Elbashir et al, 2001). The disadvantage of this approach is that silencing is dependent on the amount of siRNA administered. A sustained silencing requires a constant and expensive siRNA supply. This shortcoming is eliminated when vectors containing sequences encoding short hairpin RNAs (shRNAs) are utilized (Paddison et al, 2002). shRNAs are processed in the cells to produce siRNA. Cells transfected with these vectors can sustain RNAi-mediated gene silencing for 48 hours or longer under antibiotic selection. |
| The first large library of shRNA constructs targeting human and mouse genes was created in a retroviral vector, pShagMagic2 (pSM2) (Paddison et al, 2004). This vector is subject to frequent recombination, does not contain a GFP marker and has inefficient viral packaging that limits the use in hard to transfect cells and in vivo. |
| Aware of these problems, companies have transferred the shRNAs into more stable GFP-tagged lentiviral vectors that produce high-titer viruses (Moffat et al, 2006). Lentiviruses are suitable for transduction of hard to transfect cell lines, primary cells and in vivo applications. The latest generation of lentiviral constructs includes inducible shRNA production (TRIPZ Lentiviral Inducible shRNAmir Library®, www.openbiosystems.com). |
| All the researchers who purchased the now outdated retroviral libraries cannot take advantage of these improvements. It is very expensive for laboratories to buy a complete lentiviral library, and the single lentiviral constructs range from $209 to $428. One inexpensive way to “upgrade” the shRNA without purchasing new ones is to sub-clone them to an appropriate lentiviral vector. Open Biosystems® provides a protocol for sub-cloning shRNA constructs from pSM2 into the lentiviral vector pGIPZ but it requires expensive kits and multiple steps. Therefore, if an investigator wants to sub-clone a high number of shRNA constructs, it may be cheaper and more convenient to just purchase the constructs. |
| We developed a protocol that greatly simplifies the transfer of shRNA sequences from the retroviral pSM2 vector into the pGIPZ lentiviral vector. The improvements in the protocol are reported in Table 1. This sub-cloning protocol can be applied to sub-clone shRNA from pSM2 to newer lentiviral vectors and possibly to sub-cloning schemes that involve plasmids and fragments of the same sizes reported here. |
Materials and Methods |
| Vector preparation and restriction digestion |
| pSM2 and pGIPZ plasmids were purified with Qiagen Miniprep kits and DNA concentration was measured spectrophotometrically. All the restriction enzymes, the T4 DNA ligase and the molecular weight markers were purchased from New England Biolabs. The Open Biosystems protocols were followed for bacterial culture growth and for recombination checks. |
| pGIPZ vector (5-10μg) was digested with Mlu I/Xho I (1- 2U/μl) and the 13,087kb band was gel purified (QIAquick® gel extraction kit, Qiagen). The elution step was performed with 50μl HPLC water. The purified vector was used immediately after heat-inactivation, or stored frozen at -20oC until further use. The pSM2 vector (4μg) containing the shRNA of choice was also digested with Mlu I/Xho I (1-2U/μl) to generate a ~350bp fragment containing the shRNA, in a 20μl reaction volume (final insert concentration of 200ng/μl). After heat-inactivation (65°C for 20min) the cut pSM2 was used directly in ligation without purification. |
 |
| Ligation and transformation |
| Experimental and control ligation mixes were set up in 10μl final volume using 1:1 molar ratios of the destination vector:insert (100ng vector and 2.6ng insert). 100ng of the uncut pGIPZ vector was used as a positive control diluted in ligation buffer, 100ng of the Xho I/Mlu I pGIPZ cut vector without ligase was used to control for cutting efficiency of both enzymes and 100ng of the Xho I/Mlu I pGIPZ cut vector with 1μl of ligase were used to control for re-ligation of partially cut pGIPZ vector. In addition we had a ligation reaction without DNA added to control for DNA contamination in our reagents. |
| Ligation was performed at room temperature for 3hr to overnight and was followed by heat-inactivation at 65oC for 20min. 1μl of ligation mix was transformed in DH5α cells (InVitrogen). 300μl of the bacterial suspension were plated onto LB-ampicillin plates. Plates were incubated over night at 37oC. The next morning, colonies on the plates were counted to verify successful ligation. Typical observed colony counts were as follows: More than 100 colonies on the pGIPZ positive control and 20-100 colonies on the experimental ligation or ligations. The pGIPZ vector cut without ligase had generally 0-10 colonies while the pGIPZ cut vector added with ligase had 0-20 colonies. |
| Clone selection and verification |
| The ratio between the number of colonies on the experimental plates and the control plate (cut pGIPZ vector with ligase) was calculated and multiplied by three, the resulting number of colonies was selected for screening to validate our method. Each colony picked was dissolved in 10μl sterile HPLC water. 5μl of this suspension were used to inoculate a 5ml liquid culture of LB-ampicillin, 2μl were dotted on a master LB-Amp plate, and 2μl used as template for PCR to confirm shRNA insertion. The PCR amplification products are 600bp if the shRNA sequence is present and 500bp if it is not. The following PCR parameters were used: 5min at 94oC; then 30 cycles of, 94oC for 15sec, 56oC for 30sec, 72oC for 25sec. The pGIPZ-specific primers used were: |
| X76 Forward: 5'ACGTCGAGGTGCCCGAAGGA |
| M100 Reverse: 5'AAGCAGCGTATCCACATAGCGT |
| This PCR reaction only amplifies shRNA in pGIPZ vectors. |
| In addition to the PCR check, the pGIPZ vectors containing the sub-cloned shRNA were also digested to check for recombination of the vectors and successful insertion of the shRNA sequence. Sac II digestion was used to test for recombination of the pGIPZ vectors containing the sub-cloned shRNAs (expected bands: 7927bp, 2502bp and 1259bp). Successful ligation of the shRNA sequence was verified by excising it from the recipient pGIPZ vector using Mlu I/Xho I double digestion (expected bands: 350bp and ~10Kb). |
| Knock-down and western blotting procedure |
| SH-SY5Y cells over-expressing APP-Gal4 were transiently transfected or transduced as previously described (Zhang et al, 2007). Transfection was performed with pSM2 plasmids targeting APP or a non-silencing control (Open Biosystems), and with pGIPZ plasmids containing the same shRNA sequences as the pSM2 vectors that were sub-cloned following our protocol. Transfection was performed with Arrrest-In® (Open Biosystems®) transfection reagent. Whole cell lysates were prepared in ice-cold radio immuno-precipitation buffer (150mM NaCl, 1%, v/v, NP40, 0.5%, v/v, DOC, 1%, w/v, SDS, 50mM Tris, pH 8.0) supplemented with Halt cocktail of protease and phosphatase inhibitors (ThermoFisher). The western blotting procedure has been previously described (Zhang et al, 2007). We used anti-APP antibody clone A8717 (Sigma) and anti-actin antibody (Sigma) at 1:2000 and 1:15,000 dilution, respectively. The same cell line was transduced with viruses obtained following the packaging protocol provided by Open Biosystems® but using the same sub-cloned pGIPZ vectors previously employed for the transfection. |
Results and Discussion |
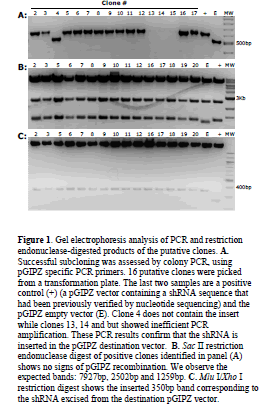
| Our protocol was successfully used to sub-clone shRNA constructs into pGIPZ vectors. As an example, one experimental transformation plate had approximately fifty colonies, the control cut pGIPZ vector with ligase had 6 colonies so the ratio of clones containing the insert to the empty ones was 50/6=8.3. Twenty-five colonies were screened with PCR using pGIPZ specific primers that amplify the shRNA sequence. In Figure 1A we show 16 of the 25 PCR reactions with the appropriate controls run on an agarose gel stained with ethidium bromide. Two clones did not show amplification (not shown), one clone resulted empty (Figure 1A; lane 4) and three clones showed suboptimal amplification (Figure 1A; lanes 13, 14, 15). All the remaining clones showed the correct band size with efficient amplification. In summary, out of the 25 colonies screened, only 3 did not show correct amplification confirming a ratio of 25/3=8.3 clones containing insert to the false positive ones. Following PCR we checked the recipient vectors for both recombination and the presence of the insert. In Figure 1B we show the results of Sac II restriction digestion of 16 clones that showed the correct amplicon following PCR. These 16 clones did not show signs of recombination. After checking for recombination of the recipient plasmid, we also checked for correct ligation of the shRNA sequence. In Figure 1C we used restriction digestion to excise the inserted shRNA from the clones and we confirmed the correct ligation in the new vector. All the clones that showed the correct PCR amplicon also showed correct restriction patterns making restriction digest unnecessary. Insertion of the shRNA sequences was also verified by sequencing. |
| In order to ensure that our sub-cloned constructs maintained silencing activity, comparable to the original pSM2 plasmids, we transfected, using the same procedure, both vectors in the same cell line. Moreover, we assessed if viral transduction of the cells also showed effective silencing. Figure 2 shows the detection of Amyloid Precursor Protein (APP) by western blotting in SH-SY5Y APP-Gal4 cells following transfection of shRNA constructs in pSM2 or pGIPZ vectors or transduction with pGIPZ containing viral particles. The silencing efficiency of the shRNA constructs in their original pSM2 vectors (Figure 2A) is comparable with the one of the shRNA subcloned in the pGIPZ vector (Figure 2B) showing that the construct was successfully inserted and does not loose targeting ability. Finally, we show that the pGIPZ construct can also efficiently decrease APP levels when delivered via viral particles (Figure 2C). |
 |
 |
Conclusions |
| The RNAi Resource Center at Drexel University purchased from Open Biosystems the pSM2 retroviral library targeting ~65,000 human genes. Our sub-cloning method has been applied successfully to ~100 shRNA sequences of interest that were transferred from the pSM2 vector into the pGIPZ empty vector. The protocol proved successful in the hands of different operators and 80% of the shRNA constructs were sub-cloned successfully on the first attempt. Unsuccessful sub-cloning was largely due to the presence of recombination in the pSM2 donor vectors. |
| pGIPZ vectors containing shRNAs obtained from our subcloning procedure produced high titer viruses (106 infectious units/ml) that were used to transduce hard-totransfect cell lines and primary neurons. We believe that our protocol will be helpful to all the investigators who have invested in the now outdated pSM2 libraries allowing them to efficiently upgrading their shRNA sequences into the new lentiviral vectors. Given the simplicity of the steps involved, it would probably be possible to implement high throughput methods to automate the rapid transfer of many shRNA simultaneously. This procedure offers approximately 60% cost savings and substantial time savings compared to the suggested protocol. |
Acknowledgements |
| The authors thank the Drexel RNAi Resource Center for providing the shRNA sequences in pSM2 vectors and the Mozino Scholarship for funding to SA. The authors also thank Molly McStravick and Nathan Miller for their technical assistance with this work. We thank Brian P Leung for technical assistance in generating the figures. This work was funded by the Drexel University, Commonwealth of Pennsylvania, the National Institute of Health (NIH) and the Alzheimer’s Association. This paper is subject to the NIH Public Access Policy. |
Compteting Interests |
| None declared |
References
|