- Journal of RNA and Genomics (2006) Research Article
Systematic analysis of the role of target site accessibility in the activity of DNA enzymes
| Graeme Doran1* and Muhammad Sohail Department of Biochemistry, South Parks Road, University of Oxford, Oxford OX1 3QU, England, UK. 1Current Address: Current Address: Department of Physiology, Anatomy and Genetics, South Parks Road, University of Oxford, Oxford, England, UK |
| Corresponding Author: Graeme Doran, Email: graeme.doran@st-edmund-hall.oxford.ac.uk, Tel: +1865 272146 |
| Received: 23 September 2014 Accepted: 11 February 2015 Published: 21 March 2015 |
Abstract
We employed an approach using oligonucleotide scanning arrays and computational analysis to conduct a systematic analysis of the interaction between catalytic nucleic acids (DNA enzymes or DNAzymes) and long RNA targets. A radio-labelled transcript representing mRNA of Xenopus cyclin B5 was hybridised to an array of oligonucleotides scanning the first 120 nucleotides of the coding region to assess the ability of the immobilised oligonucleotides to form heteroduplexes with the target. The hybridisation revealed oligonucleotides showing varying levels of signal intensities along the length of the array, reflecting on the variable accessibility of the corresponding complementary regions in the target RNA. Deoxyribozymes targeting a number of these regions were selected and tested for their ability to cleave the target RNA. The mRNA cleavage observed indicates that indeed target accessibility was an important component in the activity of deoxyribozymes and that it was important that at least one of the two binding arms was complementary to an accessible site. Computational analysis suggested that intra-molecular folding of deoxyribozymes into stable structures may also negatively contribute to their activity. 10-23 type deoxyribozymes generally appeared more active than 8-17 type and it was possible to predict deoxyribozymes with high cleavage efficiency using scanning array hybridization and computational analysis as guides. The data presented here therefore have implications on designing effective DNA enzymes.
KEYWORDS |
| Deoxyribozyme, DNA enzyme, DNAzyme, oligonucleotide array, RNA structure, cyclin B5, mfold, hybridisation |
INTRODUCTION |
| In recent years, by using in vitro selection methods, a number of short DNA sequences called DNA enzymes (or DNAzymes) have been identified that are capable of performing diverse enzymatic functions previously assigned to proteins (Breaker, 1997, 1999; Emilsson and Breaker, 2002). These functions include RNA cleavage (Breaker and Joyce, 1994; Breaker, 1997; Santoro and Joyce, 1997; Feldman and Sen, 2001), DNA cleavage (Carmi et al, 1998; Carmi and Breaker, 2001), DNA and RNA ligation (Cuenoud and Szostak, 1995; Li et al, 2000; Levy and Ellington, 2001; Flynn-Charlebois et al, 2003; Ricca et al, 2003), DNA phosphorylation (Li et al, 2000; Li and Breaker, 2001; Wang et al, 2002), DNA capping (Li et al, 2000) and peroxidase activity (Travascio et al, 1999). RNA-cleaving deoxyribozymes have been employed in a variety of in vitro applications (Stojanovic et al, 2000; Okumoto et al, 2002; Cairns et al, 2003; Mei et al, 2003; Sohail et al, 2003) and have also been shown to down-regulate endogenous and viral genes (Sriram and Banerjea, 2000; Hjiantoniou et al, 200; Fahmy et al, 2006; Zhang et al, 2006) in cell culture models and thus hold considerable potential in function analysis and gene therapies. |
| Typically, DNAzymes have a catalytic core sequence flanked by substrate-recognition ‘guide arms’ that have sequence complementary to the target nucleic acid. A critical step in the DNAzyme-mediated enzymatic processes is interaction between a DNAzyme and the target nucleic acid, RNA in the case of RNA-cleaving DNAzymes (deoxyribozymes). On binding, deoxyribozymes cleave the target RNA at a specific dinucleotide catalytic sequence (e.g., Breaker and Joyce, 1994; Santoro and Joyce, 1997; Feldman and Sen, 2001) and have been shown to catalyse RNA cleavage under a number of reaction conditions. Measuring target cleavage provides a convenient way to monitor their activity (hence their productive target binding capacity). Thus, deoxyribozymes are not only attractive anti-mRNA tools for gene silencing approaches but also present interesting models for the study of interaction between oligonucleotides and long RNAs, such as mRNAs. |
| Antisense oligonucleotides have been commonly employed in gene silencing experiments, recruiting RNase H to cleave the RNA strand of DNA:RNA hybrids. Several previous studies suggest that target site accessibility plays an important role in the activity of antisense oligonucleotides, and for efficient mRNA targeting with antisense reagents, it is important that highly accessible regions are targeted (Sohail et al, 2001; Beale et al, 2003; Bohula et al, 2003; Petch et al, 2003). |
| In the case of an oligonucleotide hybridisation with the fully complementary target results in a continuous heteroduplex. However, the two binding arms of a deoxyribozyme represent two short oligonucleotides linked through intervening sequence of the catalytic domain. Although the accessibility requirements for DNA enzymes might be expected to be similar to those of the oligonucleotides, no direct supporting evidence has yet been presented. In addition, studies investigating interactions between DNA enzymes and long RNAs are rare (e.g., Kurreck et al, 2002; Schubert et al, 2003; Schubert et al, 2004), and hence this issue requires further attention. |
| Rational design of deoxyribozyme-based gene silencing strategies require efficient methods of selecting highly active reagents. “Target scanning” is a generally used approach in which several sequential catalytic sites are targeted and deoxyribozymes are tested for their efficacy. However, unless every target site in the region of interest is laboriously tested it is quite possible to miss out the optimum target site. Another approach involves the identification of accessible sites with antisense oligonucleotides (e.g., Kurreck et al, 2002) or oligonucleotide libraries (Sohail et al, 2001) in combination with RNase H. Nevertheless, such approaches are not efficient and do not always predict active deoxyribozymes. (Cairns et al, 2000; Kurreck et al, 2002) For example, RNase H can act on short heteroduplexed regions (Wu et al, 1999) and, therefore, only partial heteroduplex formation between an antisense oligonucleotide and RNA can lead to cleavage of RNA. Such short sequences are unlikely to imitate the full length of the deoxyribozyme binding arms. Furthermore, the site of RNase H cleavage in the heteroduplex is difficult to predict and hence it may be also difficult to predict precise sequence of the accessible site (Sohail and Southern, 2000). Therefore, RNase H-based analysis alone appears insufficient for designing effective catalytic nucleic acid and may have contributed to previously reported low efficiencies of deoxyribozymes designed in this manner (e.g., Cairns et al, 2000; Cairns et al, 2003). |
| Scanning arrays provide sophisticated and powerful tools to efficiently predict accessible sites in endogenous mRNAs; antisense reagents (oligonucleotides and small interfering RNAs) designed using these arrays have been shown to work in a number of in vitro and ex vivo systems in a predictable manner (Sohail et al, 2001; Beale et al, 2003; Bohula et al, 2003; Petch et al, 2003; Sohail et al, 2005). Surprisingly, it appeared that salt concentration and presence or absence of cellular proteins had little effect on accessibility of target sites in the various mRNAs. We thus decided to use scanning arrays to conduct systematic analysis of the interaction between catalytic nucleic acids and target RNA, and investigated the usefulness of scanning arrays in designing effective deoxyribozymes. |
MATERIALS AND METHODS |
| Fabrication of the oligonucleotide array, hybridisation and image analysis |
| An oligonucleotide array was made on aminated polypropylene (Beckman Instruments, Fullerton, USA) as previously described (Sohail et al, 2001). Briefly, standard nucleotide CE phosphoramidites were used in the synthesis which was carried out on an adapted ABI 394 DNA/RNA synthesiser (Applied Biosystem). A diamondshaped mask with 42 mm diagonal was used to deliver reagents to the polypropylene surface for the in situ synthesis of the oligonucleotides. The mask was pressed against the polypropylene to create a cell. DNA synthesis reagents were introduced at the bottom of the cell and removed either from the top or from the bottom. After each nucleotide coupling, the mask was displaced by a 2.0 mm step: the result was a template comprising overlapping footprints of the mask, with each smaller diamond shape representing an individual oligonucleotide. The longest oligonucleotides were 21-mers, made along the centre line of the array. The array was deprotected in 30% ammonia solution at 55oC for 16 h in a closed chamber. |
| Hybridisation was done in 1 M NaCl, 10 mM Tris-HCl pH 7.4, 1 mM EDTA and 0.01% (w/v) SDS at 22oC for 1 hr in a rolling tube or after mounting on a glass plate as described previously (Sohail et al, 2001; Sohail et al, 2005). Autoradiography and image analysis was carried out as previously described (Sohail et al, 2001). |
| Oligonucleotide synthesis and purification |
| Oligodeoxyribonucleotides/deoxyribozymes were synthesised on an ABI394 DNA/RNA synthesiser using standard nucleotide CE-phosphoramidites (Cruachem) and were desalted through NAP-5 columns (Amersham) at least twice. Oligonucleotide stocks were maintained at 100 pmol/Nl concentration in water at –20°C. See Table 1 for a list of oligonucleotides and deoxyribozymes. |
| In vitro transcription |
| Radiolabelled transcripts for probing the scanning arrays were obtained by in vitro transcription. The template cyclin B5 plasmid was linearised with an appropriate restriction endonuclease. Transcriptions were carried out at 30°C for 90 min in the presence of 20 NCi [R-32P]UTP (<3000 Ci/mmol; Amersham), 750 NM each ATP, CTP and GTP and 18.75 NM UTP with T7 RNA polymerase (Promega). The products were purified by filtration through MicroSpin G-25 columns (Pharmacia Biotech) and analysed on a 5% (w/v) denaturing polyacrylamide gel. 5U-End-labelled transcripts for the RNase H mapping were prepared in the presence of 50 NCi [V-32P]GTP (>5000 Ci/mmol; Amersham) essentially as described above, except that the concentration of unlabelled GTP was 75 NM and that of UTP was 750 NM. |
| Deoxyribozyme cleavage assay |
| The deoxyribozymes were tested for their ability to cleave cyclin B5 mRNA under single turnover conditions in a buffer containing 10 mM MgCl2, 20 mM Tris-HCl, pH 7.5 (Kurreck et al, 2002). The 5’-end-labelled B5 mRNA transcript was mixed with the various deoxyribozymes at approximately 1:50 ratio (Kurreck et al, 2002; Schubert et al, 2004) and incubated at 30oC for 2 hr. These reaction conditions were found to be sufficient to achieve maximum cleavage of the target (data not shown). The reactions were stopped by the addition formamide gel loading buffer and the products were resolved in an 8% (w/v) denaturing polyacrylamide gel for 20-25 min. |
| Theoretical folding |
| The “bimolecular fold” function of the RNAstructure 4.3 package (Mathews et al, 2004), we used to calculate the predicted structures of deoxyribozyme hybridised to the 120 nt cyclin B5 sequence of the array, at 1M NaCl and 37oC. Folding on single deoxyribozymes was performed using the standard functions of the same package. |
RESULTS |
| Selection of deoxyribozymes |
| In order to investigate the role and the effect of target site accessibility on the activity of deoxyribozymes, accessibility profile of the target RNA was determined using a scanning array hybridisation assay (Southern et al, 1994; Sohail and Southern, 2000) since previously used approaches employing candidate oligonucleotide screening and RNase H-based assays (Cairns et al, 2000; Kurreck et al, 2002) appeared insufficient for such analysis. Scanning arrays provide an exhaustive accessibility profile of the target sequence and are, therefore, powerful tools for such a study (Sohail et al, 1999). Based on the accessibility predictions using these arrays, highly efficient antisense reagents have been designed for in vitro, ex vivo and in vivo applications (Sohail et al, 2001; Beale et al, 2003; Bohula et al, 2003; Petch et al, 2003; Sohail et al, 2005). |
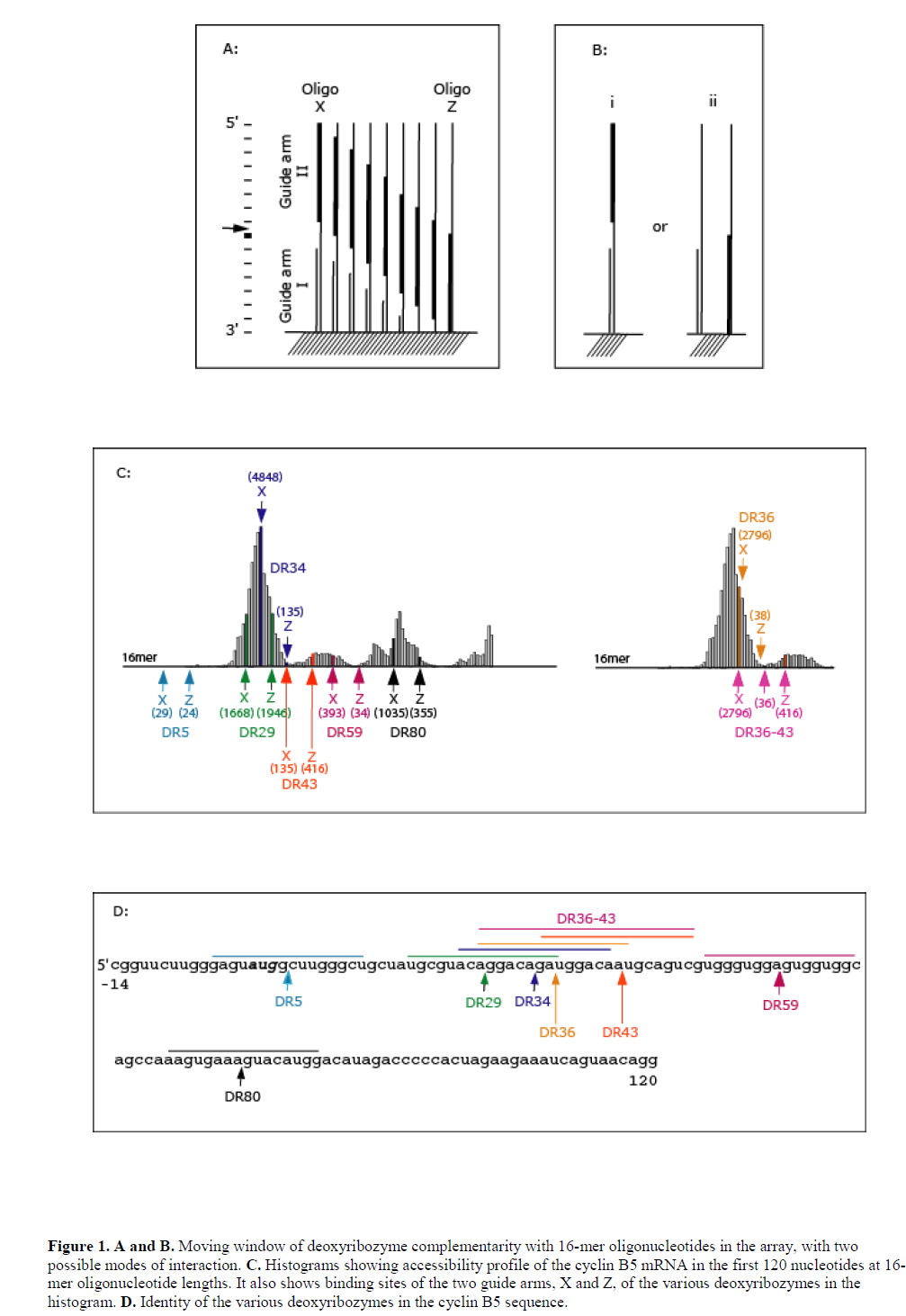
| A scanning array against cyclin B5 mRNA of Xenopus laevis was fabricated by in situ oligonucleotide synthesis on the surface of aminated-polypropylene (Sohail et al, 2001) (also see Materials and Methods). The array represented complements of the first 120 nt of the coding sequence and consisted of oligonucleotides ranging in size from monomers to 21-mers. Radio-labelled B5 mRNA transcript was hybridised to the array and image analysis revealed accessibility profile of the target mRNA (see Figure 1C). We then identified catalytic sites for ‘10-23’ and ‘8-17’ deoxyribozymes within this region and based on the accessibility profile designed a number of deoxyribozymes containing both 8-17 and 10-23 catalytic motifs targeting both accessible and inaccessible regions (see Table 1 and Figure 1C). Hybridisation potency of the 16-mer antisense oligonucleotides in the array to B5 mRNA transcript was used as guide to designing deoxyribozymes, since 16-mers represented the total sequence length covered by the two guide arms of the deoxyribozymes. The hybridisation potencies of 16-mer oligonucleotides (and hence target site accessibility) varied along the length of the array and, in many cases, even few base shifts had considerable effect on binding. It was not possible to use hybridisation potencies of 7-mers in the design, since these oligonucleotides showed very little hybridisation in the array (data not shown). The most likely reason for this lack of hybridisation is that such short sequences do not form stable heteroduplex under these experimental conditions (see Mir and Southern, 1999). |
 |
| Deoxyribozymes DR1 and DR5 targeted inaccessible sites, while DR29, DR34, DR36, DR36-43, DR43, DR59 and DR80 targeted sites with varying degree of accessibility (Figure 1C). DR36-43 had two catalytic domains separated by a 6 nt sequence complementary to the target. Both arms of the deoxyribozymes DR1 and DR5 were complementary to inaccessible sites in the mRNA. DR29 and DR80 were complementary to accessible 16-mer sequences and independently both their guide arms were also complementary to individual accessible 16-mer sequences. DR34, DR36, DR43 and DR59 targeted accessible 16-mer sequence; however, individually at least one of their arms was complementary to an inaccessible 16-mer sequence. |
| We wanted to explore the role of target site accessibility in predicting the activity of a particular deoxyribozymes. It was of interest to note whether the accessibility to either the 5’ or 3’ guide arms of the deoxyribozyme might impart greater activity, i.e., the two guide arms behave as two independent oligonucleotides (Figure 1A and B). If the deoxyribozyme behaved as a single oligonucleotide, then activity would be expected to relate to the accessibility of an oligonucleotide sequence complementary to both the binding arms. If, however, the binding arms hybridise as independent oligonucleotides then the accessibility of either arm would allow cleavage at the target site, the binding of an individual arm either affecting the local RNA structure and enhancing accessibility of the RNA sequence complementary to the second arm, else enhancing the local concentration of the complementary sequence and improving the chance for productive binding. |
| Cleavage efficiencies of deoxyribozymes – the effect of target site accessibility |
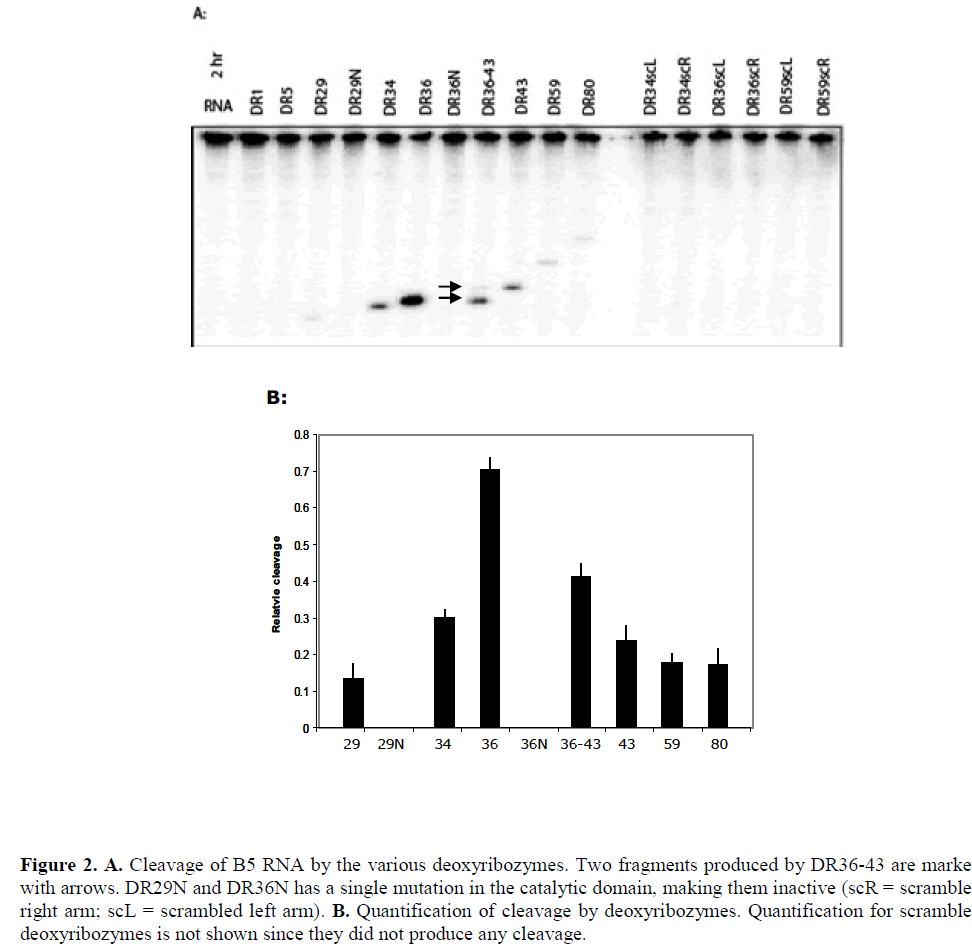
| The results of cleavage of B5 mRNA by the various deoxyribozyme are shown in Figure 2. Deoxyribozymes DR1 and DR5 targeted inaccessible sites in the target and, as expected, did not show any detectable cleavage of the transcript while all others cleaved B5 mRNA to a varying degree, with DR36 producing the highest cleavage. Deoxyribozymes DR29, DR34 and DR36 targeted the same highly accessible region of the transcript but cleaved the transcript to different extent: while both binding arms of DR29 were located within accessible regions, at least one of the two arms of DR34 and DR36 was positioned in an inaccessible region. The relative accessibility of the deoxyribozymes dinucleotide target site as predicted by the array (that is, the hybridisation intensity) was a superficially poor predictor of deoxyribozyme activity, as the most accessible target site (DR29) was much less active than the comparable 8-17 deoxyribozyme DR34. |
| Logically, hybridisation of either or both 7-mer guide arms is more likely to correctly orientate the deoxyribozyme for target site cleavage than chance hybridisation at the target dinucleotide site. Moreover, Ota et al (Ota et al, 1998) showed that the helical structure of the hybridised guide arms affects the activity of the deoxyribozyme. We decided to focus on the accessibility and character of the guide arm sequences, to analyse the role of these parameters in predicting deoxyribozyme activity. |
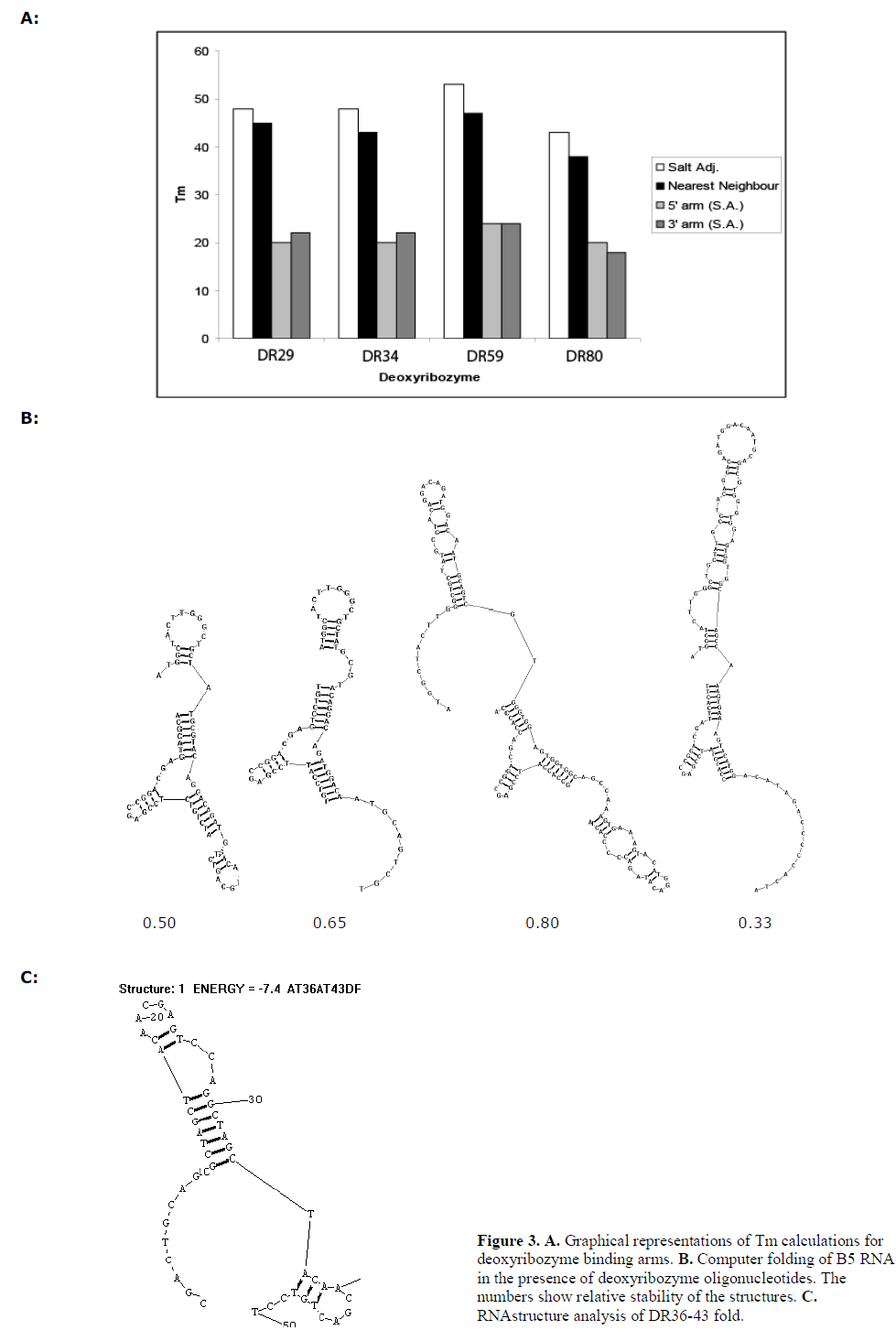
| We focussed upon the 8-17 deoyribozymes to study this. Nearest-neighbour Tm calculations were performed for the sequences of each guide arm, and for both guide arms combined (data presented in Figure 3A). |
| Using the “bimolecular fold” function of the RNAstructure 4.3 package (Mathews et al, 2004), we calculated the predicted structures of deoxyribozyme hybridised to the 120 nt cyclin B5 sequence of the array. The 8-17 deoxyribozyme is predicted to adopt a common fold when fully hybridised to the target sequence (Figure 3B) and this was taken to represent the “active” conformation. RNAstructure can predict multiple folds in order of descending thermodynamic stability. We tallied the number of “active” fold configurations predicted by within 20% of the most stably predicted conformation (usually ~4kjmol-1). This set of folds was taken to represent the most common configurations of the hybridised deoxyribozyme allowing for thermal fluctuations (data presented in Figure 3B). |
| The hybridisation intensities of the peaks representing the guide arms of the deoxyribozymes were also analysed, either as an average of the 5’ and 3’ guide arm accessibility, solely 5’ or 3’ accessibility, or as the most accessible guide arm. The order of accessibility, and therefore activity, was compared to that predicted by computational folding or Tm calculation. The order of activity predicted by the array was 34>29>80>59. Tm calculations predicted 59>34>29=80, whereas RNA folding suggested 59>34=29>80. The actual activity observed was of the order 34>59>29>80, which was not predicted by any of the analytical methods. |
| We performed the same analysis for two 10-23 deoxyribozymes. DR43 was predicted most active by computational methods, whereas DR36 was targeted a more accessible sites on the array array, and ultimately showed greater cleavage activity in vitro. |
| Although none of the methods presented proved entirely accurate, it was possible to draw some information from them. Both computational analyses identified a stable interaction of DR59 with the target sequence, due to a high GC content of the site. Although this site is quite inaccessible as predicted by the array, DR59 binds with strong affinity, and so, will likely result in productive cleavage whenever the site is accessible for binding. Array accessibility correctly predicted the relative activity of deoxyribozyme targeted to more accessible sites, but with lower binding affinity. This suggests that both guide arm sequences and target structure play a role in predicting the activity of a deoxyribozyme. |
 |
 |
| Effecter oligonucleotide provides additional evidence for the role of target site accessibility |
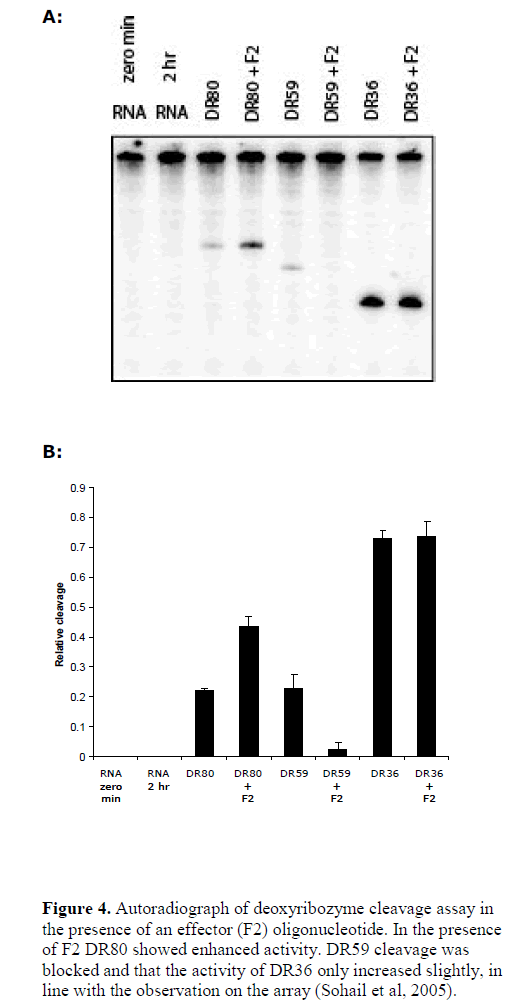
| We further tested the role of RNA target accessibility by the application of an effecter oligonucleotide (Sohail et al, 2005). Using B5 mRNA as target, we previously found that the binding of an oligonucleotide (effecter F2) produced dramatic changes in the structure of target RNA and lead to considerable increase in the accessibility of a neighbouring sequence (Sohail et al, 2005). Coincidently, the binding site of DR80 fell within the region showing increased accessibility. This offered a good model system to test whether increasing or decreasing accessibility of the same target site would also alter the activity of the deoxyribozyme in parallel. We performed a cleavage assay in the presence of the effecter oligonucleotide F2. As controls we also tested two other deoxyribozymes: DR59 whose binding site overlapped with that of F2 and DR36, which had binding site in the region that showed only minor increase in accessibility in the presence of F2 (see Figure 4). The results show that indeed in the presence of F2 DR80 showed enhanced efficiency, the activity of DR59 was blocked and that the activity of DR36 only increased slightly, in line with the observation on the array hybridisation (Sohail et al, 2005), and suggesting target site accessibility to be one of the most important factors in determining deoxyribozyme activity. |
| Such array-based approaches also offer a rational way of designing effecter oligonucleotides (Wang et al, 2002; Schubert et al, 2004; Sohail et al, 2005) to enhance deoxyribozyme activity. In the previous studies, Depankar Sen’s Laboratory developed effecter oligonucleotides that made a three-way junction with the deoxyribozyme and the target and proposed their use in therapeutic applications(Wang et al, 2002). However, their tests were based on the use of short RNA targets which is unrealistic for gene silencing applications and their attractive approach needs further validation with long RNA targets (such as mRNAs). In mRNAs the regions of accessibility are generally short and limited in length and thus designing such three-way effecters may be challenging. With the array-based approach it is possible to select effectors that are most likely to enhance deoxyribozyme activity. Such effecter mediated enhancement of deoxyribozyme activity may be useful where a specific sequence of interest is to be targeted that is otherwise poorly accessible. |
 |
 |
| Deoxyribozyme guide arms recruit RNase H for target cleavage |
| DR36 was the most active deoxyribozyme identified by scanning array, and presents a good candidate for further studies ex vivo. The cellular environment is far more complex than that of the in vitro assay, however, and we wanted to explore whether the deoxyribozyme guide arms could recruit RNase H to cleave the mRNA target at sites within the guide arms. A deoxyribozyme cleavage assay was performed with the addition of RNase H (Figure 5). In the presence of RNase H, cleavage of the RNA target occurred at the dinucleotide specifically targeted by the deoxyribozyme, and also at the adjacent guide arms. This observation suggests that guide arms can recruit endonucleases to sites of just 7 base complementarity. Since it is unrealistic to expect a 7-mer sequence to be unique within the genome when designing a deoxyribozyme, steps must be taken to ensure that guide arms to not induce endonuclease mediated cleavage, such as 2’O-Methyl ribonucleotide modification of these sequences. Additionally these potential “off-target” effects of deoxyribozymes, due to binding of either guide arm to a RNA must be considered when evaluating cell culture results, preferably through genome-wide array analysis of mRNA expression. |
| Dual-domain Deoxyribozymes – specifying distance between cleavage sites |
| We examined the effect of combining DR36 and DR43 since they were both active single deoxyribozymes and were spaced by a moderate six nucleotide spacer sequence. The resultant deoxyribozyme, DR36-43, contained two 10- 23 catalytic domains and three guide arms. Both domains cleaved that target (Figure 2A) but to a lesser extent than that by the two enzymes DR36 and DR43 separately. RNA folding analysis indicated that the longer dual-domain deoxyribozyme oligonucleotide might form more stable internal structures that inhibit the binding to the target site (Figure 3C), and therefore lowering the activity of the two individual domains. |
| Although in this particular case we did not see a positive effect of the inclusion of two domains, careful engineering of a dual domain enzyme might be useful in certain applications that demand length specific cleavage, for example the generation of short RNAs from larger precursors. |
DISCUSSION |
| There are several factors that may affect the activity of antisense oligonulceotides and deoxyribozymes, and these factors must be weighed in the rational design of deoxyribozymes against long target RNAs. Particular success has been achieved in modifiying deoxyribozymes with either 2’OMe ribonucleotides, or Locked nucleic acid (LNA) monomers that enhance the oligonucleotide affinity for the target RNA (Schmidt et al., 2004). This approach validates the importance of binding affinity (measured as Tm) in deoxyribozyme design. Accessibility of a sequence is a pre-requisite for binding, and so accessibility must also be taken into account when optimising deoxyribozyme design, and accessibility profiling has been proven effective in designing antisene reagents in a number of other systems (Sohail et al, 2001; Beale et al, 2003; Bohula et al, 2003; Petch et al, 2003; Schubert et al, 2004). We set out to compare computational methods and in vitro accessibility profiling to determine the usefulness of both in designing deoxyribozymes. |
| Though theoretical folding is often believed not to be accurate in predicting structure of long RNAs (Sohail et al, 1999; Ho et al, 1998), it is likely to produce correct folding of short sequence. We folded deoxyribozymes using the RNAstructure package (http://rna.urmc.rochester.edu/rnastructure.html) (Mathews et al, 2004) together with a 120 nt sequence corresponding to the first 120 nt of the cyclin B5 mRNA. It is not possible to study interaction of DNA with RNA or vice versa - the program will only model both DNA or both RNA sequences as input. Since deoxyribozymes are short, we decided to use their homologous RNA sequence as input instead of DNA in the analysis, since we did not find significant difference in the folding or DNA or RNA sequences corresponding to deoxyribozymes. However, in the case of target RNA sequence, we preferred to use RNA since its DNA homologue had a very different fold. Although this is not the ideal situation it is the best that the computer analysis offers and further developments in the algorithms are likely to improve such studies. The stability of the structure was judged by the number of correctly folded structures present amongst those predicted. The rational for this is that a “molten” RNA will adopt several low energy structures as it fluctuates. If the majority of these structures contain a deoxyribozyme in the active form then cleavage is more likely to occur. The order of activity predicted by this method was essentially equivalent to the Tm analysis - 59>34>29>80. Array hybridiasation predicted an order of activity of 34>29>80>59, and the actual activity observed was 34>59>29>80. Clearly both methods are able to predict the general order of relative activity, although DR59 defeats the analysis. |
| DR59 is targeted to a relatively inaccessible region, yet appears to form a stable interaction with the target site which overcomes this. To show that enhancing accessibility can enhance activity we employed an effecter oligonucleotide to disrupt the structure around the DR80 binding site, to make it more accessible. The resulting cleavage is greater by a factor of 2 in the presence of the effecter, confirming that target site accessibility is a contributing factor. |
| Accessibility profiling predicted deoxyribozyme activity based upon the maximally accessible guide-arm, rather than specifically the 5’ or the 3’ guide-arm. Noticeable also was the potential for non-specific off-target cleavage indicated in Figure 5. Both of these results suggest that care must be taken that partially complementary sequences are not inadvertently targeted for cleavage. Modifications that asymmetrically enhance the binding affinity of one arm over the other, or inhibit endonuclease mediated cleavage, might be employed to limit off-target binding in cases where complementarity cannot be avoided. |
| The results presented here indicate that target site affinity is an important contributor to deoxyribozyme activity. However, thermodynamic calculations alone cannot predict deoxyribozyme activity in silico and accessibility profiling is necessary to predict the most active deoxyribozymes in vitro. The design of optimally active deoxyribozymes must therefore focus upon the RNA target structure to the same degree as chemical modifications of the deoxyribozyme when designing the best candidates for catalytic nucleic acid therapeutics. |
ACKNOWLEDGMENTS |
| This work was funded by the Medical Research Council, UK. The authors thank Professor Sir Edwin Southern for providing laboratory facilities to carry out this work. |
STATEMENT OF COMPETING INTERESTS |
| The authors declared no competing interests. |
SHORT COPYRIGHT STATEMENT |
| This is an open access article, published under the terms of the Licence for Users available at http://www.libpubmedia.co.uk/ RNAiJ/LicenceForUsers.pdf. This licence permits noncommercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
References |
|