Research Article - Biomedical Research (2017) Volume 28, Issue 2
In-silico designing of novel inhibitors of rho-GTPase activating protein for treatment of glaucoma
Li Yin, Jing Zhu, Xun Bao, Yong Yao*Department of Ophthalmology, Wuxi People’s Hospital Affiliated to Nanjing Medical University, Wuxi, Jiangsu Province 214023, China
- *Corresponding Author:
- Yong Yao
Department of Ophthalmology
Wuxi People’s Hospital Affiliated to Nanjing Medical University, China
Accepted on July 05, 2016
Abstract
Increased intraocular pressure resulting in the optical neuropathy characterised by permanent blindness is called glaucoma. The aqueous humor accumulates in the posterior chamber because of the slower release rate of aqueous humour as compared to the rate of its formation results in increased intraocular pressure, which creates pressure against the optic nerve causing the death of nerve cells. In 2010 more than 60.5 million individuals are affected by glaucoma and it is also the worldwide leading cause of irreversible blindness. Multiple population-based surveys estimated that about 80 million peoples would be affected by the year 2020. Earlier it is shown that the in-vitro and ex-vitro inhibition of Rho pathway by specific inhibitors causes the contractile tone relaxation of the cells which are actively involved in the aqueous outflow pathway, thus reduces intraocular pressure by increasing aqueous outflow. Thus in this research paper in-silico techniques are used to develop Rho GTPase protein inhibitors for treatment of glaucoma and it is predicted that ZINC17465958 and ZINC05124957 molecules are potent inhibitors of the Rho GTPase protein. These molecules will prove to be promising lead compound for further structure based discovery of drugs for treatment of glaucoma as they are having very good pharmacokinetic properties without presence of any toxic effects.
Keywords
Glaucoma, Docking, Virtual-screening, Ligand, Drug design.
Introduction
The optic neuropathy caused by a group of ocular diseases characterized by the loss of visual sensitivity and field associated with the changes in the optic nerve head are called glaucoma. Traditionally it is diagnosed by intraocular pressure (IOP), which has an important role in its pathogenesis, but is no longer a diagnostic criterion for glaucoma nowadays. Glaucoma may remain undiagnosed mostly in more than half of the patients because of its initial asymptomatic symptoms at its early stages in developing and developing countries of the world [1,2].
Increased intraocular pressure is one of the most common causes for the glaucoma, which is caused by the improper draining of the aqueous humor produced in the eye chambers present between the cornea and the lens by the eye itself. The intraocular pressure between 10 to 21 mm Hg is considered to be normal and increased intraocular pressure above this range creates pressure against the optic nerve resulting in the death of nerve cells. Aqueous humor is formed in the epithelial layer of the ciliary body by both filtration and secretion. The aqueous humor formation by ultra-filtration is influenced by the change in intraocular pressure, blood pressure and pressure gradient. The epithelial cells present in the ciliary body actively secretes sodium and bicarbonates and other solutes such as ascorbates into the aqueous humor, resulting in the movement of water from ciliary stromal ultra-filtrate pool to the posterior chamber for the formation of aqueous humor [2,3].
The Rho GAP pathway has a crucial role in the modulation of cells of the cytoskeletal integrity, the synthesis of extracellular matrix which is an important component of the tissues responsible for the outflow of the aqueous humor as well as the regulation of the endothelial cells permeability of the Schlemm's canal. The trabecular meshwork (TM) contracts because of the activation of Rho/ROCK pathway and its inhibition results in the relaxation of TM with increased outflow facility and subsequent decrease in intraocular pressure (IOP) [4]. The specific cause responsible for the glaucomatous optic neuropathy is presently unknown and traditionally, the increased IOP was considered to be the only main cause responsible for this damage and it is believed that the outcome of these processes results in the apoptosis of the retinal ganglion cells, which causes the axonal degeneration, and finally permanent loss of vision. Although IOP poorly predicts which patients will have visual field loss, the risk of visual field loss clearly increases with increasing IOP within any range. In glaucoma multiple mechanisms are involved simultaneously and are responsible for the cellular death of the retinal ganglion and their axons. Astrocytes and other cells in the optic disk supportive matrix are highly sensitive to pressure and may produce changes and remodelling of the disk, resulting in axonal death. Vasogenic theories suggest that optic nerve damage results from insufficient blood flow to the retina secondary to the increased perfusion pressure required in the eye, dysregulated perfusion, or vessel wall abnormalities, and results in degeneration of axonal fibres of the retina [2].
The cellular interaction with their environment is controlled by Rho pathway by regulating various integral intracellular processes. The Rho super family consists of three subfamilies- Rho, Rac, and Cdc. The microtubular dynamics, actin cytoskeleton, vesicle trafficking, cellular polarity and progression of cell-cycle are regulated by Rho proteins. The Rho pathway expresses human trabecular meshwork and having an important role in the regulation of the cellular morphology and contractile tone involved in the aqueous outflow pathway. A lipophilic isoprenoid group is present in the Rho proteins, which is used for their attachment into the cellular membrane and essentially important for performing biological functions. The inhibition of the HMG-CoA conversion to L-mevalonic acid results in the inhibition of the synthesis of isoprenoid intermediaries, including FFP and GGPP. Because of the localisation and trafficking inhibition of these GTPase proteins at subcellular level, some significant functional changes are observed. Importantly, the partial function of the post-translationally immature forms of Gproteins is maintained and the activity of mature membraneanchored proteins is interfered [1].
The inhibition of Rho and its downstream effectors proteins like Rho kinase (ROCK) and Rho GTPase alters the contractile properties of the conventional outflow pathway. Rho signalling is responsible possessing a contractile tone in the cells of the conventional pathway. The phosphorylation of ROCK inhibits the myosin-binding subunit present in the myosin light chain (MLC) phosphatise resulting in the increased MLC phosphorylation and myosin contractility, thus the stress fibres production and focal adhesion rate is increased. Earlier it is shown that the in-vitro and ex-vitro inhibition of Rho pathway by specific inhibitors results in relaxation of the contractile tone of cells of the aqueous outflow pathway. This increases aqueous outflow and reduces intraocular pressure [5,6].
About 10 percent peoples of 70 million of peoples affected by glaucoma worldwide are believed to be bilaterally blind. After cataract, glaucoma is the second leading cause of blindness globally according to the data obtained from World Health Organization (WHO). Maximum number of glaucoma patients is found in China followed by Europe/US and India. The individual of any age group, i.e. newborn, infants, children or elderly people may easily get affected with Glaucoma. In 2010 more than 60.5 million individuals are affected by glaucoma and it is also the worldwide leading cause of irreversible blindness. Multiple population-based surveys estimated that about 79.6 million individuals would be affected by the year 2020 [7-9]. Because of the continuous increase in the glaucoma patients globally, there is an urgent need to develop a potent drug for its treatment. In this research paper in-silico approaches are applied to develop a novel Rho GTPase Activating Protein's inhibitor as a novel drug for treatment of glaucoma. The three dimensional model of crystal structure of a small G protein in complex with the GTPase-activating protein rhoGAP (PDB id-1AM4), obtained from RSCB Protein Data Bank is used for experimental studies for this research paper [10-12].
Materials and Methods
All the molecular docking simulations were carried out utilizing softwares Raccoon, AutoGrid4 and AutoDock 4 [13].
Selection and preparation of protein
The human Rho GTPase activating protein bound with its endogenous ligand phosphoamino phosphonic acid-guanylate ester (PDB id-1AM4) was downloaded from RCSB protein data bank. The 1AM4 protein complex consists of six chains: A, B, C, D, E and F. Chain D of the protein were kept for molecular docking studies while other chains were deleted by using the Chimera software. Similarly, chain D was selected out of three pair of homologous chains in protein 1AM4. The proteins preparation for molecular docking simulation is performed by removal of unwanted water molecules from protein, removal of ligand from active ligand binding site of protein and addition of polar hydrogen's to the protein. Amino acids GLY515, LYS516, THR517, THR535, GLY560, GLN561, ASP618 and LEU660 were involved in the binding of the ligand to the active binding site of the protein 1AM4.
Selection of chemical libraries
In-silico virtual screening was performed with "NCI Diversity Set II” having 1880 diverse ligand molecules to identify potent lead molecules for the protein 1AM4. All the ligands used in virtual screening process follows Lipinsky’s rule of five, i.e. having physicochemical parameters in the given range of ClogP<5, H bond donor<5, H bond acceptor<10, Molecular weight<500 etc.
Grid box formation
Three dimensional grid boxes covering the ligand, all the active ligand binding residues and ligand binding site, by placing the box in the centre of the ligand is used for performing molecular docking simulation. The grid parameters used in this experimental study are described in Table 1. These grid parameters are utilized for all the docking runs.
| Proteins | x-D | y-D | z-D | Spacing (?) | x center | y center | z center |
|---|---|---|---|---|---|---|---|
| 1AM4 | 40 | 40 | 40 | 0.464 | 5.594 | -4.929 | -10.576 |
Table 1. The coordinates of grid box for the three proteins.
Grid maps preparation
The Autogrid utility of the AutoDock suite is used to generate map files for different atom types present within the ligands and the receptor. In current experimental study the map files for the following atoms i.e. A, C, OA, HD, F, I, Br, Cl, N, NA, SA, S etc. were prepared by Autogrid. The map files generated by the Autogrid are further utilized by the AutoDock for performing molecular docking simulations.
Docking parameters
Primary conformational search approach Lamarckian genetic algorithm (LGA) of AutoDock suite is utilized for performing molecular docking simulation [13]. LGA creates a trail population of various possible conformations, and these conformations compete in a similar manner that of biological evolution for selection of the conformation having lowest binding energy among the successive generations by mutating conformations and exchanging conformational parameters. The additional feature of LGA is the conformational search and their local space for the individual conformations by finding local minima and passing this information to later generations. The binding free energies of small molecules to the target macromolecules were predicted by using semi-empirical free energy force field, which is based on the comprehensive thermodynamic model which allows intra-molecular energies incorporation into the predicted binding free energy by evaluating bound state energy with the unbound state energy. For performing molecular docking simulation of each ligand a docking parameter file was prepared by using following docking parameters, i.e. 150 GA runs, 250000 maximum number of evaluations, 27000 maximum number of generations and 0.02 rate of gene mutation.
Docking method validation
The conformational orientations and position of the ligand identified by the molecular docking simulation study represents valid and reasonable modes of binding of the inhibitors. The various docking parameters applied here were validated by re-docking crystallized ligand, i.e. phosphoamino phosphonic acid-guanylate ester over Rho GTPase activating protein of Homo sapiens. The docked conformation of the bound ligand is overlayed over the crystallized chemical structure to calculate a root mean square deviation (RMSD) value.
Virtual screening process
Raccoon software is utilized for the preparation of all the necessary files for performing in-silico virtual screening process. Molecular ligand library containing number of ligands is splits into individual ligand file and converted to the pdbqt format necessary for autodock with the help of Raccoon. It also filters the ligand molecules which are not following some necessary rules for pharmacokinetic profiling such as Lipinski's rules, fragment-like rule of 3, and drug-likeness etc. Input file is validated at every step by checking for the presence of non- standard atom types and ensuring that parameters, input filenames, and grid maps have a coherent format [13-19].
Result analysis of docking simulation
After the completion of the virtual screening technique, a script summarize_results.py released by Scripps Research Institute was used for the extraction and sorting out the binding energies of the docked ligand and which is further utilized for the selection of the best hit molecules. The evaluation of the results obtained after the molecular docking simulation were performed on the basis of hydrophobic and polar interactions obtained between ligand and active binding residues present in the ligand binding site of the macromolecule and the empirical range of the binding energy must comes in between -5 to -15 Kcal/Mol. Binding affinity is calculated from the formula:
Ki=e[(ΔG/(RT)]
Where, ΔG=change in free energy upon binding, R=gas constant and T=temperature.
Prediction of ADME & toxicity of lead compounds
The presence of any toxic effects and pharmacokinetic profiling in the lead molecules selected after performing the virtual screening technique were evaluated by using the online program OSIRIS Molecular Property Explorer. This program predicts the presence of major toxicities like mutagenicity, tumorigenicity, irritant effect and reproductive effects within the lead molecules on the basis of functional group present in their chemical structure. This program also calculates druglikeness and drug score of the lead molecule on the basis of its physicochemical properties which governs their pharmacokinetic profile [20-23].
Results and Discussion
Table 2 depicts the results of the internal validation for the proteins 1AM4.
| Proteins | Interacting residues | Internal validation RMSD value | Binding energy (Kcal/Mol) | Binding affinity (nM) |
|---|---|---|---|---|
| 1AM4 | GLY515, LYS516, THR517, | 0.45 | -9.71 | 104.08 |
| THR535, GLY560, GLN561, | ||||
| ASP618, LEU660 |
Table 2. Internal validation results for the three proteins.
The Figure 1 illustrates the binding mode and chemical interactions of the ligand phosphoamino phosphonic acidguanylate ester within the active binding site of human Rho GTPase activating protein. These observed results demonstrate that we are exactly simulating the process which is involved inside the human body by using in-silico molecular docking simulation technique, i.e. we have observed similar binding interactions involved in the binding of the docked ligand with the receptor protein which are already present in the bioactive crystallized structure obtained by the wet lab experimental studies.

Figure 1: Binding mode and chemical interactions of the ligand phosphoaminophosphonic acid-guanylate ester within the active binding site of human Rho GTPase activating protein.
While Figure 2 shows the overlay of chemical structures of docked conformation of the ligand with respect to its crystallized bioactive conformation. The perfectly overlayed docked conformation demonstrates that the ligand is binding exactly in the similar manner as that of bioactive conformation of the ligand present in the downloaded structure model of the crystallized structure obtained from the RSCB protein data bank.

Figure 2: Overlay of chemical structures of docked conformation of the ligand with respect to its crystallized bioactive conformation.
Visual analysis of the interactions observed between ligand and protein for top ranking poses for each ligand is performed for selection of the potential ligands. The results obtained after performing the virtual screening technique for human Rho GTPase activating protein are shown in Table 3. Table 4 illustrates that all the top ten hits following Lipinski’s rule of five.
| S. No. | Compound ID | Chemical Structure | Binding Energy (kcal/mol) | Binding Affinity (µM) |
|---|---|---|---|---|
| 1 | ZINC03954520 | 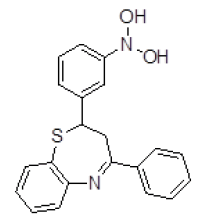 |
-10.64 | 15.89 |
| 2 | ZINC04376856 | 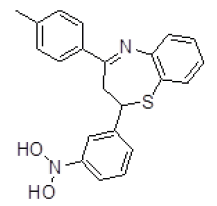 |
-10.08 | 40.57 |
| 3 | ZINC29590300 | 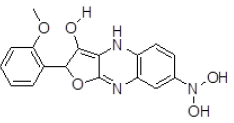 |
-9.15 | 197.61 |
| 4 | ZINC01588812 | 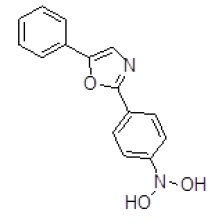 |
-9.14 | 201.24 |
| 5 | ZINC01045089 | 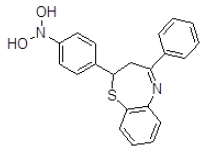 |
-9.12 | 206.91 |
| 6 | ZINC17465958 | 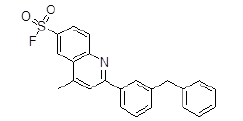 |
-9.11 | 208.31 |
| 7 | ZINC29590296 | 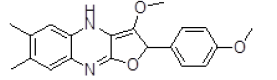 |
-8.94 | 279.02 |
| 8 | ZINC05479118 | 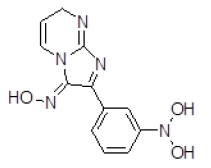 |
-8.89 | 302.36 |
| 9 | ZINC01753336 | 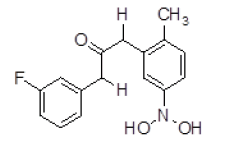 |
-8.88 | 311.26 |
| 10 | ZINC05124957 | 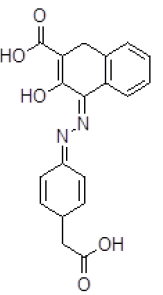 |
-8.88 | 309.54 |
Table 3. Binding energy and affinity of top five hits from "NCI Diversity Set II” after virtual screening on human Rho GTPase activating protein.
| S. No. | Compound ID | Mol. Wt. | ClogP | 2D PSA (Å2) | HBA | HBD |
|---|---|---|---|---|---|---|
| 1 | ZINC003954520 | 362.1 | 4.27 | 56.06 | 4 | 2 |
| 2 | ZINC04376856 | 376.12 | 4.79 | 56.06 | 4 | 2 |
| 3 | ZINC29590300 | 341.1 | 1 | 106.78 | 7 | 4 |
| 4 | ZINC01588812 | 268.1 | 1.86 | 69.73 | 4 | 2 |
| 5 | ZINC01045089 | 362.1 | 4.27 | 56.06 | 4 | 2 |
| 6 | ZINC17465958 | 391.1 | 6.43 | 47.03 | 3 | 2 |
| 7 | ZINC29590296 | 336.14 | 3.7 | 52.08 | 4 | 1 |
| 8 | ZINC05479118 | 273.1 | 0.17 | 104.25 | 8 | 3 |
| 9 | ZINC01753336 | 289.11 | 2.87 | 60.77 | 4 | 2 |
| 10 | ZINC05124957 | 352.1 | 2.16 | 119.55 | 7 | 3 |
Table 4. “Lipnski’s rule of five” for the hits on human Rho GTPase activating protein.
The top ten ligand molecules out of large number of ligand molecules present in the molecular library are short listed on the basis of the lowest binding energy obtained after performing the virtual screening technique. The ligand-protein interaction between the bound ligand and the protein macromolecule present in 1AM4 were compared with respect to the compounds which are short listed by the virtual screening technique. The identification of the best poses were performed using these criteria in their given order of preferences, i.e. lowest binding energy in the largest sized cluster, number of hydrogen bonds formed with the active binding residues and conservation of the interactions those were observed during control docking. These steps were followed to ensure that the hits were actually interacting in the defined ligand binding site present in the macromolecule.
The short listed ligand molecules by virtual screening technique are further evaluated for their pharmacokinetic profiling and presence of any toxic effects by using various software's and online web-servers like Marvin, Osiris molecular property explorer etc. After pharmacokinetic profiling and toxicity evaluation it is found that all the short listed ten molecules are having very good pharmacokinetic properties and two lead molecules ZINC17465958 and ZINC05124957 are free from the presence of any toxic effects.
Out of the top 10 lead molecules selected after performing the virtual screening technique, two lead molecules ZINC17465958 and ZINC05124957 shows highly promising results as it shows potent inhibition of the Rho GTPase protein with good pharmacokinetic properties without any toxic effects. Very good drug-likeness and Drug Score values were observed for both the lead molecules ZINC17465958 and ZINC05124957. The ADME profiling and toxicity estimation results of all the selected lead molecules obtained after performing virtual screening technique are shown in Figure 3.

Figure 3: ADME-T Profiling and toxicity prediction of virtually screened lead molecules.
Conclusion
Molecular docking simulation based in-silico virtual screening was carried out using Autodock4 was very useful in short listing potential lead compounds. The molecular docking simulation based virtual screening technique is proved to be very useful in short listing of potential lead molecules out of large number of ligands present in the molecular ligand libraries. Two compounds ZINC17465958 and ZINC05124957 shows highly promising results with potent inhibition of the Rho GTPase protein as well as having very good pharmacokinetic properties without presence of any toxic effects. In particular, these two molecules serves as a promising lead compound for further structure based discovery of drugs for treatment of glaucoma.
References
- Rao VP, Epstein DL. Rho GTPase/Rho kinase inhibition as a novel target for the treatment of glaucoma. Bio Drugs 2007; 21: 167-177.
- Wang SK, Chang RT. An emerging treatment option for glaucoma: Rho kinase inhibitors. ClinOphthalmol 2014; 8: 883-890.
- Padmanabhan PP, Rao PV. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. Am J Physiol Cell Physiol 2010; 298: C749-C763.
- Dipiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM. Pharmacotherapy- A Pathophysiologic Approach. McGraw-Hill, New York, 2016.
- Pokrovskaya1 O, Wallace D, O’Brien C. The Emerging Role of Statins in GlaucomaPathological Mechanisms and Therapeutics. Open J Ophthalmol 2014; 4: 124-138.
- Feng Y, LoGrasso PV, Defert O, Li R. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J Med Chem 2016; 59: 2269-2300.
- World Health Organisation. Vision 2020:The Right to Sight. Global initiative for the elimination of avoidable blindness. Action Plan 2006-2011, World Health Organisation.
- Leonord R. Statistics on Vision Impairment: A Resource Manual. April 2002, 5th Edition.
- Solomon A. Magnitude and Causes of Childhood Blindness and Severe Visual Impairment in Sekoru District of Jimma Zone, South West Ethiopia: The Key Informant Method. Project Report, London School of Hygiene & Tropical Medicine. 2008-09.
- Wang J, Liu X, Zhong Y. Rho/Rho-associated kinase pathway in glaucoma (Review). Int J Oncol 2013; 10: 1357-1367.
- Tuccinardi T, Cascio MG, Di Marzo V, Manera C, Ortore G, Saccomanni G, Martinelli A. Structure-based virtual screening: identification of novel CB2 receptor ligands. LettDrig Design Discov 2007; 4: 15-19.
- Von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 2007; 6: 967-974.
- Forli Stefano.; Raccoon AutodockPrepration Tool; The Scripps Research Institute; 2009.
- Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. Structural characterization of the 1918 influenza H1N1 virus neuraminidase. J Virol 2008; 82: 10493-10501.
- Mihajlovic ML, Mitrasinovic PM. Another look at the molecular mechanism of the resistance of H5N1 in?uenza A virus neuraminidase to oseltamivir. BiophysChem 2008; 136: 152-158.
- Avian influenza (bird flu): an introduction. Newsletter: Division of Communicable Disease Control WHO. Nov 2005.
- Phosrithong N, Ungwitayatorn J. Molecular docking study on anticancer activity of plant-derived natural products. Med Chem Res 2009; 19: 817-835.
- Verma RP, Hansch C. A QSAR study on influenza neuraminidase inhibitors. Bioorg Med Chem 2006; 14: 982-996.
- De Clercq E, Neyts J. Avian influenza A (H5N1) infection: targets and stratgeis for chemotherapeutic intervention. Trends PharmacolSci 2007; 28: 280-285.
- Furuta Y, Takahashi K, Kuno-Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. Mechanism of action of T-705 against influenza virus. Antimicrob Agents Chemother 2005; 49: 981-986.
- Phoristhong N. Molecular docking study on anticancer activity of plant-derived natural products. Med Chem Res 2009; 19: 817-835.
- George PA, Edmond LJ. Bioisosterism: A rational approach in drug design. Chem Rev 1996; 96: 3147-3176.
- Hansch C, Garg R, Kurup A. Searching for allosteric effects via QSAR. Bioorg Med Chem 2001; 9: 283-289.