- Journal of RNA and Genomics (2005) Review Article
Designer siRNAs to overcome the challenges from the RNAi pathway
| Sumedha D Jayasena* Amgen Inc., Oncology Research, One Amgen Center Drive, Thousand Oaks, CA 91320, USA |
| Correspondence to: Sumedha D Jayasena, Email: sumedhaj@amgen.com, Tel: +805 447 5627, Fax: +805 499 2751 |
| (Received 13 October 2005; Revised 17 November 2005; Accepted 21 November 2005, Available online 30 November 2005; Published 28 February 2006) |
Visit for more related articles at Journal of RNA and Genomics
Abstract
Small interfering RNA (siRNA)-based technology is playing a pivotal role in understanding gene function. Huge siRNA libraries coupled to high-content screening are being applied to decipher molecular circuitries, as well as to identify novel therapeutic targets. Further, the technology is finding its way towards therapeutic applications. In the midst of all this excitement, the siRNA technology is faced with challenges, arising mostly from siRNAs being a nucleic acid molecule, and also from the baggage it inherits from the RNA interference (RNAi) pathway, which is critical to the function of siRNAs.
Keywords |
| RNAi, siRNA, microRNA, gene silencing, off-target effects, siRNA design, siRNA delivery, shRNA expression, immune activation |
Introduction |
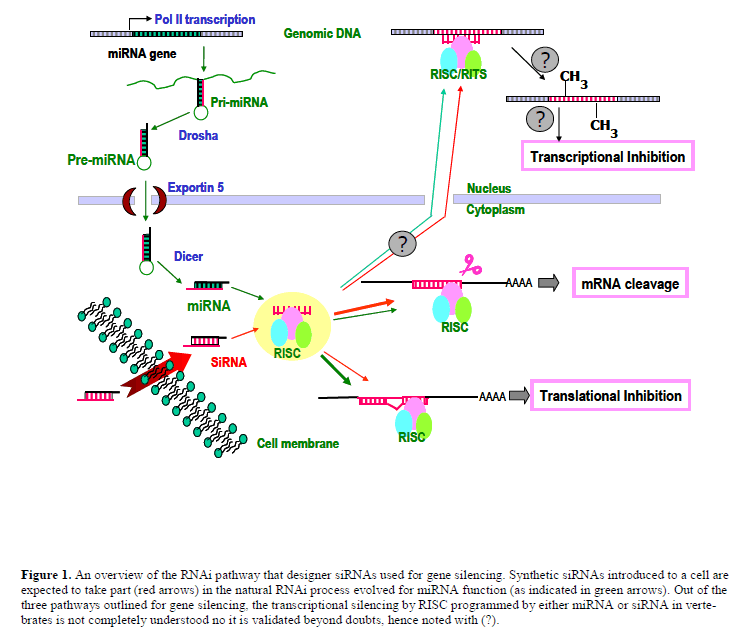
| Within a short span of time after its discovery (Caplen et al, 2001; Elbashir et al, 2001), siRNAs have become the key technology in the application of reverse genetics to understand gene function by eliminating or reducing the expression of a gene of interest. The technique is easy to use, relatively specific, and highly-efficacious with broad applicability across multiple cell types in human. siRNAs exert their function by high jacking an evolutionary conserved cellular process called RNA interference (RNAi). RNAi, thought to have been evolved as a defence mechanism in primitive organisms (Lu et al, 2005; Plasterk, 2002; Wilkins et al, 2005; Zamore, 2002), plays a pivotal role in gene regulation in both plants and animals by using an endogenous, non-coding small RNA species called MicroRNAs (miRNA). At a fundamental level, RNAi is a sophisticated antisense mechanism (Figure 1) that captures one strand from a double stranded miRNA (or siRNA) into an effecter protein complex called RISC (RNA-induced silencing complex). The RISC complex uses the captured strand as a ‘guide’ to target complementary sequences within an mRNA. While the target sites for miRNAs are typically located within the 3’-untranslated regions (3’- UTRs), siRNAs are designed to target anywhere within an mRNA. The fate of the mRNA is dictated by the degree of complimentarity between the guide strand of the siRNA duplex and the target region; if the complimentarity is perfect or near perfect (as in the case of siRNAs) the endonuclease activity within the RISC cleaves the target mRNA; however, if the complimentarity is imperfect (as with most miRNAs), a suppression of protein translation occurs through a mechanism that is poorly understood even todate. Both mechanisms down regulate protein expression, but the action of the one that utilizes incomplete base pairing is reversible, as it does not permanently reduce the level of the target mRNA. |
| In this review, keeping in mind the potential of siRNA technology to become a therapeutic modality, I have made an attempt to highlight challenges that the technology is currently facing. These challenges are stemming from the ability of the exogenous siRNA to hijack a natural molecular pathway, evolved and optimized mostly for miRNAs, and siRNA being a nucleic acid molecule. As challenges bring new opportunities, I have included recent advancements in the field to bring the siRNA technology a step forward in realizing its therapeutic potential. |
Challenges Arising From The RNAi Pathway |
| An exogenous siRNA is a molecular mimicry of the enzyme product of natural double-stranded RNA that enters |
| RNAi pathway. It is also designed to have a full complimentarity to the target site of interest to ensure long-term gene silencing through mRNA cleavage. Early successes of gene silencing by siRNAs based on simple design rules (Elbashir et al, 2002) lost consistency when applied to diverse gene sets (Aza-Blanc et al, 2003). Later, some understanding of the mechanism of the RISC assembly improved siRNA design (Khvorova et al, 2003; Schwarz et al, 2003). In the natural RNAi pathway, in almost all cases, only the miRNA strands (miRs), but not their compliments (miR*s) are incorporated into the RISC. The analysis of miRNA duplexes revealed an asymmetry at the two ends dictated by the base pairing stability (Khvorova et al, 2003). Analogous asymmetry with less stable 5’end of the antisense strand compared the 5’end of the sense strand was also noticed among highly-functional siRNAs (Khvorova et al, 2003; Schwarz et al, 2003). This differential thermodynamic stability of the two termini of an RNA duplex dictates which strand gets incorporated into the RISC. Hence, by design, the preferred antisense strand can be directed to populate the RISC, ensuring efficient degradation of target mRNA in the absence of potential off target silencing mediated by the sense strand. While this discovery made a significant advance towards the improvement of siRNA design, specific base preferences at certain locations within an siRNA duplex have also been noted (Reynolds et al, 2004), and incorporated into design algorithms. A phosphate group at the 5’end of the antisense strand is required for efficient RNAi (Nykanen et al, 2001; Martinez et al, 2003). The core activities of the RISC appear to reside within a complex containing a singlestranded guide RNA and an Argonaute family protein (Liu et al, 2004; Meister et al, 2004). The crystal structures of the PIWI domain of an Argonaute protein revealed the existence of a highly basic pocket chelated with a metal ion to capture the charged 5’phosphate group of the guide strand (Ma et al, 2005; Parker et al, 2004). This interaction may facilitate the positioning of the guide strand for effective target cleavage by “slicer” endonuclease activity residing in the human Argonaute2 (Ago2) protein, as well as to increase the half-life of the RISC. Hence, the elimination of the 5’phosphate group in the sense strand by chemical modification would exclude the participation of the sense strand in unintended gene silencing. |
 |
| A multiple number of algorithms have been developed for picking effective siRNAs (Gong and Ferrell, 2004; Reynolds et al, 2004; Snove et al, 2004), including a patternlearning approach based on neural networks (Huesken et al, 2005). To improve specificity, these algorithms perform in silico screening to eliminate candidate siRNAs with near perfect pairing to unintended genes in the human (target) genome. In addition to the focus on the siRNA duplex, the attention to the target mRNA accessibility has also been shown to be important for efficient gene silencing (Bohula et al, 2003; Heale et al, 2005; Overhoff et al, 2005; Schubert et al, 2005). An empirical approach to probe accessible sites within a target mRNA (Heale et al, 2005) didn’t gain a widespread use due to laborious nature of the technique, especially in high throughput applications. However, accessibility of target sites needs to be addressed to further optimize siRNA design rules. One approach would be to screen regions of potential mRNA target sites by an RNA folding program such as mfold (Zuker, 2003) or sfold (Ding et al, 2004) to eliminate sites within tightly folded RNA motifs that may resist targeting by the RISC. This may not be 100% effective in eliminating all inaccessible sites due to either the potential longrange interactions within long mRNA sequences or sequestering of target sites upon interaction with RNAbinding proteins or both. |
| A potential pitfall in current siRNA designs is the difficulty in weeding out sequence-specific off-targets resulting from the pairing rules identified for miRNA/target recognition. The 5’seed region that defines 2-7 nts of a miRNA mediates target recognition (Lewis et al, 2003), allowing multiple targets to be regulated by a single miRNA. This promiscuous target recognition characteristic of miRNAs has a toll on siRNA applications. Microarray analysis indicated that siRNAs could mediate gene silencing of unintended targets through their 5’regions (Jackson et al, 2003). In general, gene silencing mediated by siRNA through miRNA mechanism is not transparent at the mRNA level, but detectable at the protein level. This phenomenon, already noticed in siRNA applications (Jackson et al, 2003; Saxena et al, 2003), would become more wide-spread as investigators start looking into the global changes of the proteome affected by siRNA experiments (Scacheri et al, 2004) or the use of a large siRNA libraries on a single gene (Lin et al, 2005). It is needless to emphasize the value of siRNA libraries designed to knockdown many gene families, and their applications in identifying novel gene targets for therapeutic interventions, as well as in deciphering molecular pathways. However, a caution must be exercised in interpreting the results derived from genome wide or gene familywide siRNA screening in the absence of extensive validation in subsequent experiments to rule out potential off targets leading to inadvertent erroneous conclusions. Keeping the concentration of siRNA at relatively low, yet effective, level is important to avoid non-specific gene regulation mediated by high concentrations of siRNA (Persengiev et al, 2004; Semizarov et al, 2003). On the other hand, the elimination of the off-target silencing mediated by the 5’seed pairing is extremely challenging, and most siRNAs will have a number of unintended targets affected by 5’seed pairing, as a six contiguous string of bases occurs quite frequently within a genome (once in every 4 kb sequence). |
| More than one siRNA to a given target is commonly used in discovery research to ensure a single phenotype resulting from on-target gene silencing by most siRNAs used. “Multiple siRNAs per target” approach is acceptable for discovery research but fails in potential therapeutic applications. Ideally, therapeutic applications demand ontarget mechanism with little or no off-target silencing – a formidable task. However, if the number of potential offtargets can be reduced to a minimum level, then therapeutic siRNAs may not be all that different from a small molecule kinase inhibitor that inhibits multiple kinases. The latter class of molecules is beginning to enter into clinical trials. Nonetheless, further refinement of design algorithms would be warranted for siRNAs especially aimed for therapeutic developments. A significant progress has been made in developing algorithms for predicting miRNA targets based on the 5’seed pairing (John et al, 2004; Kiriakidou et al, 2004; Stark et al, 2003), the same exact mechanism that renders siRNAs for off-target recognition. In the future, it is expected to merge miRNA target prediction algorithms with those developed for siRNA design to eliminate candidate siRNAs with potential off target gene silencing through the undesirable miRNA-related mechanism. |
| Recently, another twist to the already complex subject of off target gene silencing was introduced by demonstrating transcriptional gene silencing by siRNAs (Kawasaki and Taira, 2004; Morris et al, 2004). The siRNA-guided RISC apparently silenced the transcription of specific chromatin regions consisting of homologous DNA by recruiting factors that methylate genomic DNA. Both siRNA and miRNA guided RISC have been shown to be present and active in both cytoplasmic and nuclear compartments (Robb et al, 2005). Whether the partitioning of the RISC between nucleus and cytoplasm is a consequence of the degradation of nuclear membrane during cell division or through an active transport mechanism is not currently understood. In spite of some experimental evidence, it is worth noting that the entire phenomenon of transcriptional silencing by RISC (or RITS: RNA-induced transcriptional silencing) in vertebrates is controversial. If this phenomenon is proven to exist, then strategies to keep this activity at bay would be important for siRNA applications. |
Challenges From Sirna Being a Nucleic Acid Molecule |
| Elicitation of immune response in mammals |
| Microbes trigger innate immune response in mammals by activating Toll-like receptors (TLR). Among others, the molecular species that are sensed as non-self by TLRs include ssRNA, dsRNA and CpG motifs in DNA derived from viruses, bacteria and fungi. The innate immune response triggered by TLRs includes the activation of Type 1 Interferon (IFN) system. In addition, the dsRNA also triggers other antiviral responses comprising of the activation of PKR, Oligoadenylate synthase and RNase L (Katze et al, 2002). However, in general, siRNAs tend to escape the activation of antiviral response that is commonly trig gered by dsRNA molecules longer than the canonical siRNAs. This view held initially on siRNA (Elbashir et al, 2001) has recently been challenged, as some siRNA molecules elicit interferon response in cultured cells and in animals (Bridge et al, 2003; Judge et al, 2005; Kariko et al, 2004; Kim et al, 2004; Sledz et al, 2003). Recently, specific molecular footprints within siRNAs that could stimulate undesirable immune response have been discovered (Hornung et al, 2005; Judge et al, 2005). These studies identified two different sequence elements (5’-UGUGU-3’ and 5’-GUCCUUCAA-3’) with a common function: exerting immune response when delivered using cationic lipids, which directs siRNAs to the endosomal pathway where they encounter TLRs. The elimination of such putative immune stimulatory elements (ISEs) from an siRNA sequence reduced the immune response when delivered in liposome formulations both in vitro and in vivo settings. Surprisingly, the siRNA molecules that lacked the known putative ISEs were also capable of eliciting immune response in mice (Morrissey et al, 2005), suggesting the possible existence of hitherto unknown ISEs in siRNA. Most importantly, chemical modifications introduced at different positions of siRNAs led to a marked reduction of inflammatory cytokines induced in mice. This suggests that the chemical modification of siRNAs may be a potential approach for evading mammalian immune surveillance evolved to sense unmodified natural nucleic acids. Interestingly, the aforementioned two ISEs were identified from studies on a rather limited sequence space. It is likely that new ISEs in siRNA would be identified in the future by using random sequence libraries of siRNAs exhausting all possible nucleotide combinations. The elimination of all possible potential ISEs in combination with specific chemical modifications would expect to provide cleaner siRNA molecules for future therapeutic applications. |
| In vivo systemic delivery – another major challenge to be defeated |
| In spite of certain shortcomings associated with the technology, siRNA has become the laboratory workhorse for elucidating gene function. Yet the technology failed to penetrate into therapeutic arena for the foremost challenge in developing effective strategies for in vivo delivery. The initial proof-of-principal experiments demonstrating in vivo efficacy in mice were based on an unorthodox delivery approach of high-pressure tail vein injection (HTVI), a method inappropriate for human. Recently, several groups have devised different approaches for systemic in vivo delivery of siRNAs (Figure 2). Such approaches would provide a foundation on which further advancements to be made in the future to make siRNA therapeutics a reality. HTVI delivery of naked siRNAs did not show in vivo immune response in mice (Heidel et al, 2004), but siRNAs formulated into cationic lipids and delivered by low pressure IV did trigger an in vivo immune response (Judge et al, 2005), suggesting siRNAs complexed with lipids traffic through cellular compartments involved in immune activation. |
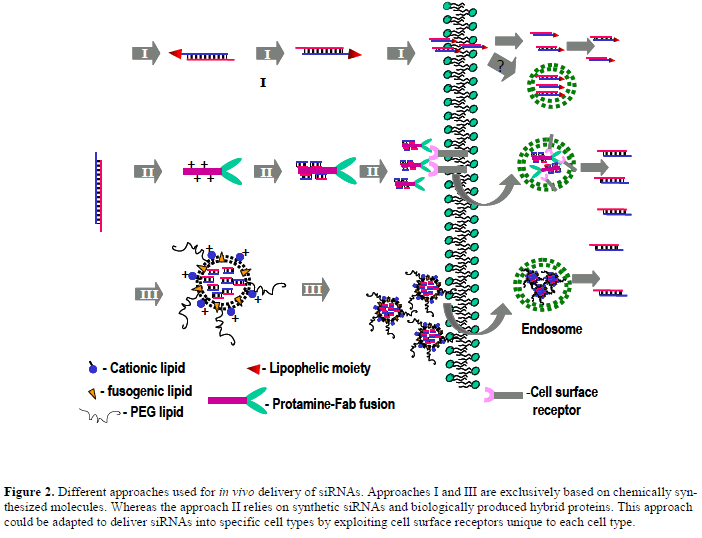 |
| The direct conjugation of cholesterol to the 3’end of the sense strand has improved in vivo delivery of chemically modified naked siRNAs by intravenous injection (Soutschek et al, 2004). Cholesterol-conjugated, chemically modified siRNAs against the liver target Apo-B showed reduction of the cognate protein level in mouse plasma by 30-70% (depending on the siRNA used), when administered at a relatively high dose (50 mg/kg) for three consecutive days and assayed a day later. Cholesterol conjugation appears to facilitate intracellular uptake of siRNA in tissue culture in the absence of a lipid-based transfection agent. When delivered in vivo, it significantly enhanced pharmacokinetics parameters, slowing down the plasma clearance presumably upon binding to serum proteins. |
| A novel approach (Song et al, 2005) took the advantage of a chimeric molecule made up of two molecular modules; one for cellular homing, provided by heavy-chain antibody fragment (Fab), and the other for nucleic acid loading, provided by a positively charged peptide derived from basic protein protamine that condenses nucleic acids. This approach also allowed for the delivery of naked siRNAs avoiding the use of cationic lipid formulations. Using an anti-HIV-1 envelope Fab fused to protamine peptide showed uptake of fluorescein-tagged siRNA specifically into HIV-1 envelope expressing cells in culture. The use of anti-gag siRNA in this system showed inhibition of viral replication in HIV-infected primary T-cells, cells that generally resist transfection even with widely used lipid formulations. The cell-specific delivery was extended to in vivo settings using a model in which a melanoma cell line engineered to express HIV-1 envelope protein implanted into the flanks of mice. The animals were challenged with a cocktail of siRNAs against c-myc, MDM2 and VEGF genes loaded onto anti-HIV-1- Fab-Protamine fusion construct by either intratumor or intravenous injections on 0, 1 and 3 days after implanting tumor cells (approximately 4 mg/kg of siRNA per injection). Intra-tumoral delivery showed higher efficacy than the intravenous delivery in reducing tumor volume up to 9 days after treatment. These preliminary results are encouraging and warrant further investigations in robust animal models in which the regression of already established tumors could be addressed. To allow such studies the authors have taken initial steps in making a fusion construct of anti-ErbB2-Fab-Protamine to target ErbB2- expressing breast tumors. Again, encouraging results showing specific targeting of ErbB2-expressing MCSF7 cells were obtained in culture. Though this is an interesting approach for in vivo delivery of siRNA, additional research and improvements are necessary before the technique is embraced widely. Some of these issues may include improving the payload capacity of siRNAs, as each complex tend to carry approximately six siRNA molecules in the current format; demonstrating specific delivery into a wider tissue/cell types using a variety of receptor-based affinity tags; evaluating pharmacokinetics and dynamics of these complexes, shelf life and their overall stability, as well as possible unanticipated toxic effects such as immunogenecity; and finally, the production cost associated with a protein-RNA complex of this nature for therapeutic intervention in human. |
| An integrated approach that incorporated refined siRNA picking that avoided potential ISEs, chemical modifications that enhanced stability and programmed RISC assembly, and formulation into specific liposome particles comprising of cationic, fusogenic lipids and an external coating with a hydrophilic polyetheleneglycol has been recently reported (Morrissey et al, 2005) and showed notable improvement in possible therapeutic delivery. In this example, antiviral siRNAs carrying positional chemical modifications, encapsulated into specialized liposomal particles demonstrated dose-dependent reduction of viral DNA (> 1 log) seven days after the last dose of IV delivery (3 or 0.3 mg/kg for 3 consecutive days) in a mouse model of HBV replication. Based on highly-effective gene silencing at <10 nM siRNA in tissue culture experiments, this is a reasonable dose for in vivo efficacy. The mouse liver model of HBV is established by HTVI of a HBV vector 6 days prior to the delivery of encapsulated siRNAs, a potential caveat associated with the interpretation of the delivery efficacy into an already compromised mouse liver. This has been somewhat addressed by extending weekly maintenance dose up to 6 weeks after the initial dose, and still showing significant reduction of serum HBV DNA titer. Further investigations into a possibility of silencing an endogenous gene in mouse liver in the absence of a prior HTVI would be useful in determining the full potential of this approach. |
| While broad therapeutic applications prefer nonviral delivery of siRNAs to avoid obvious complications associated with viral delivery into human, in vitro applications certainly benefited from the development of plasmid-based vectors that are designed to deliver siRNA through shRNA (small hairpin RNA in which the siRNA forms the basepaired stem). The initial excitement in siRNA technology led to discover ways to deliver siRNA into cells using expression vectors. Based on the small size of shRNAs, RNA polymerase III (pol III)-based expression vectors were developed (Paddison et al, 2004) (Tuschl, 2002), as promoters that recruit pol III efficiently transcribe short non-coding RNA species like tRNAs and small nucleolar RNAs. The development of pol III-based shRNA expression systems was greeted with much enthusiasm, as they were readily adapted to viral delivery systems that expanded the number of cell types amenable for gene silencing. Conditional or regulated gene silencing provides a window of opportunities to study genes that can be difficult to address otherwise, including those that regulate cell cycle, apoptosis and proliferation. Further, long-term silencing of genes may lead to conditions that are nonphysiological. Pol II-based promoters have already been engineered to provide conditional gene expression in response to small molecules such as tetracycline or ecdysone. To afford conditional shRNA expression, inducible pol IIIbased systems were subsequently developed (Czauderna et al, 2003; Gupta et al, 2004; van de Wetering et al, 2003). |
| All this development took place before the elucidation of miRNA biogenesis to the current understanding. Interestingly, miRNAs are encoded in long transcripts derived from pol II transcription and efficiently processed into a small fragment of ~22-nt prior to get incorporated into the RISC (reviewed in Bartel, 2004; Kim, 2005). Furthermore, miRNAs can function as siRNAs and vice versa (Doench et al, 2003; Zeng et al, 2002; Zeng et al, 2003). Signals that are required for efficient miRNA processing have been shown to reside within less than 40 nts either side of a miRNA precursor (Chen et al, 2004) and would be important for efficient gene silencing when shRNA sequences were embedded in place of a miRNA. Some miRNAs are naturally expressed in a single transcript in a multicystronic fashion. Embedding shRNAs designed to silence multiple genes within a single pol II transcript would allow concurrent silencing of multiple genes. Previously developed regulatable pol II promoters could now be adaptable for conditional gene silencing using transcripts based on miRNA frameworks (Stegmeier et al, 2005), leaving behind pol III-based expression systems developed exclusively for shRNA expression. In a way, the development of shRNA expression vectors have come a full circle. |
Realizing Therapeutic Potential Of siRNA |
| The full potential of siRNA therapeutics would be realized only after a solution to an effective intracellular delivery is achieved. While systemic applications of siRNA to combat multiple disease areas wait for such a breakthrough, applications in which local delivery of siRNA (or shRNA) into restricted tissue compartments have advanced much closer to human therapeutics. Clinical trials are currently underway in patients with macular degeneration to control retinal angiogenesis by intraocular delivery of synthetic siRNA to VEGF receptor (http://www.sirna.com). A lentiviral vector-based delivery of shRNA to control HIV replication is being developed as a therapy for AIDS lymphoma patients (http://www.benitec.com). Clinical trials will soon be initiated in this patient population by ex-vivo transduction of autologous hematopoietic progenitor cells with viral vectors carrying an arsenal of shRNA constructs. The results of these clinical trials would be critical for the field of siRNA therapeutics and will pave the way for diverse applications in the future. |
Conclusions and Perspective |
| The siRNA technology has already become an indispensable tool for understanding the roles of a myriad of genes identified by recent sequencing efforts of a number of genomes including the human. To that end, high-throughput genome wide RNAi combined with high-content screening approaches are already being developed (Brummelkamp et al, 2003; Wheeler et al, 2005). These efforts will lead to uncover novel biological pathways, as well as to identify missing links in already known cellular circuitries (MacKeigan et al, 2005; Willingham et al, 2004). Further, the indirect impact of this powerful technology in developing novel therapeutics will soon come into life by identifying and validating novel therapeutic targets that has not been able to accomplish so quickly by using alternate approaches. Synthetic lethal screening in tissue culture is just one such approach with the potential for identifying novel therapeutic targets (Brummelkamp and Bernards, 2003; Wheeler et al, 2004). |
| However, the technology has failed to deliver its promise on siRNA as a broad therapeutic modality for overwhelming challenge associated with the systemic in vivo delivery of nucleic acid molecules, albeit examples of local siRNA delivery has reached human clinical trials. This has been the case in the past for all other nucleic acids based technologies, including antisense, ribozymes and triplexes. Another parallel to antisense technology is the identification of immune stimulatory motifs in siRNA sequences, similar to those discovered in antisense molecules (Krieg et al, 1995). However, various chemical modifications developed for other nucleic acids based therapeutic approaches are beginning to permeate into the siRNA field to improve the potential of siRNA therapeutics. In this light, a designer therapeutic siRNA molecule (Figure 3) would |
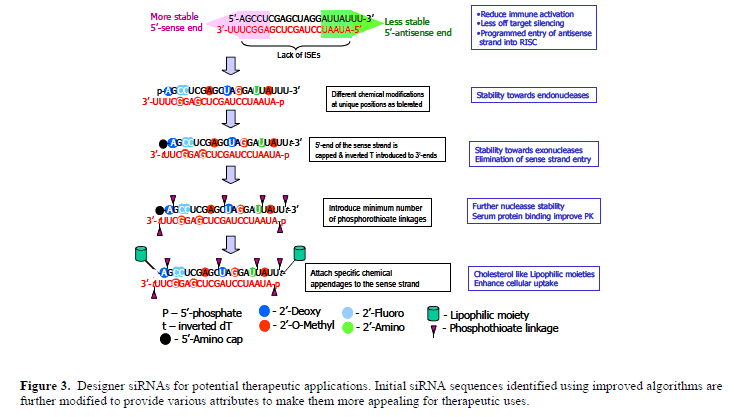 |
| carry various chemical modifications at defined positions for: protection from nucleases and to modulate interactions with serum proteins to improve in vivo pharmacokinetics; guiding the antisense strand into the RISC assembly to improve efficacy to lower the therapeutic dose; blocking the sense strand participating in RNAi to eliminate off target gene silencing; eliminating immune activation by evading TLRs; attaching other molecular appendages (for example, cholesterol) to improve in vivo, as well as tissuespecific delivery. Some of the chemical modifications can be generalized as they can be introduced independent of the siRNA sequence, but certain others will be sequencespecific. The latter class of modifications requires a careful study with each siRNA sequence to identify how exactly the various modifications introduced at each position would affect critical parameters like the immune activation and RISC assembly. The development of in vitro assays that will allow effective screening of a host of chemical modifications deemed necessary. A recent report indicating the successful in vitro reconstitution of the RISC activity with recombinant hAgo2 (Rivas et al, 2005) would be useful in that regard. |
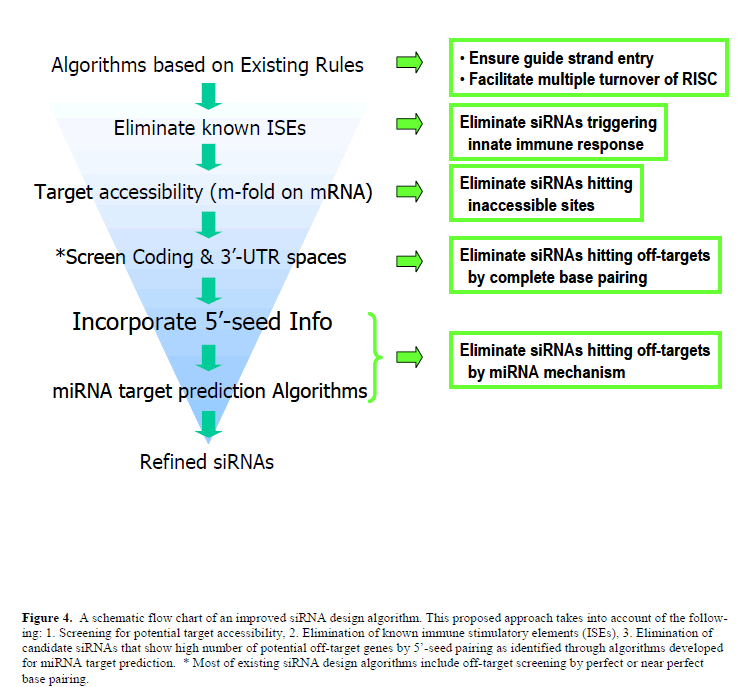
| Off-target gene silencing, which could be managed for in vitro applications could still be an issue for therapeutic applications, and requires creative approaches to reduce the number of potential off targets. Future siRNA design algorithms (Figure 4) that incorporate steps to eliminate known ISEs as well as the miRNA target prediction algorithms would be of value to significantly reduce the number of potential off targets for a designer siRNA. In spite of all these developments that are important for therapeutic applications, the real success of siRNA therapeutics will depend on the advancement made on efficient in vivo systemic delivery of siRNA molecules, along with the outcomes of the ongoing and planned clinical trials. Based on already made progress in this area, it is reasonable to anticipate an optimistic future for siRNA therapeutics. |
 |
Statement of Competing Interests |
| The author declared no competing interests. |
List of Abbreviations |
| miRNA; MicroRNA |
| RISC; RNA-induced silencing complex |
| TLR; Toll-like receptors |
| ISE; Immune stimulatory element; |
| HTVI; High-pressure tail vein injection |
References |
|