- Journal of RNA and Genomics (2005) Research Article
Design and application of a versatile expression vector for RNAi in mammalian cells
Visit for more related articles at Journal of RNA and Genomics
Abstract
Here we report a versatile mammalian expression vector called pRIGHT11 for production of small interfering RNA (siRNA) in cells. This vector uses opposing eukaryotic RNA polymerase III promoters U6 and H1 to drive the expression of short siRNA. We have demonstrated the effectiveness of pRIGHT11- generated siRNA in sequence-specific inhibition of transfected reporter genes and endogenous genes. Furthermore, this retrovirus-based vector can carry a random library of siRNA, and thus can be applied to rapid screening of novel genes involved in specific cellular responses.
Keywords |
| RNAi, retroviral, polymerase III, Smad, vector, delivery, siRNA, shRNA |
Introduction |
| The genomic databases have become available for human and a few other model organisms. In these databases, the number of genes of unknown functions has been growing exponentially. The traditional genome-wide methods for analyzing gene functions include genetic screens for phenotypic alterations in model organisms. Although these methods have been extremely productive in determining gene functions, they often involve use of expensive, tedious and lengthy procedures including chemical or molecular mutagenesis, chromosomal walking, and introduction of DNA libraries. These limitations hinder the use of genetic analysis to study gene functions in mammalian systems in a high-throughput manner. |
| Recently, long double stranded RNAs (dsRNA) were used for genome-wide screens in C. elegans to study its developmental processes (Ashrafi et al, 2003; Kamath et al, 2003) and in Drosophila cells to dissect cellular pathways (Kiger et al, 2003; Lum et al, 2003). Since dsRNA triggers RNA interference (RNAi), these early studies demonstrated the great potential of RNAi to study gene function in a genome-wide fashion. RNA-mediated interference (RNAi) is a evolutionarily conserved mechanism for inhibiting expression of specific genes by dsRNAs (for recent reviews see Denli and Hannon, 2003; Dykxhoorn et al, 2003; Tuschl, 2003; Voorhoeve and Agami, 2003). However, the dsRNA approach cannot be used in mammalian cells, because dsRNA induces a strong antiviral response resulting in cell death. Tuschl and colleagues cleverly showed that 21-23 bp long small interfering RNAs (siRNAs) can elicit RNAi in mammalian cells without inducing antiviral responses (Elbashir et al, 2001). siRNAs can be introduced into cells as RNA (which is either chemically synthesized or in vitro transcribed) or expressed from short-hairpin RNA (shRNA)-encoding DNA expression vectors (for a review see Wilson and Richardson, 2003). shRNA expression vectors often harbor an RNA polymerase III promoter, such as H1 or U6 promoter, that drives the expression of a shRNA (Brummelkamp et al, 2002; Paddison et al, 2002; Sui et al, 2002). Large-scale siRNA screens can be carried out in mammalian cells, as a recent effort identified new players in death receptor-mediated apoptosis (Aza-Blanc et al, 2003). However, such screens require synthesis of a large number of siRNA in order to cover the whole genome. Otherwise, the screens are biased toward the selected siRNAs of chosen gene targets. Ideally, a random library of siRNA will serve the purpose for a genome-wide screen. |
| In this study, we describe an siRNA expression system that uses two polymerase III promoters that are arranged to direct the expression of both sense and antisense strands of siRNA in an opposite orientation. This system has considerable potential for the rapid identification of functional genes in a genome-wide genetic screen. Here we report that this retrovirus-based RNAi vector could efficiently suppress the expression of reporter and natural genes making it possible to use this system as a mammalian genomic tool in genome-wide screens. |
Materials And Methods |
| Molecular cloning and pRIGHT11 construction |
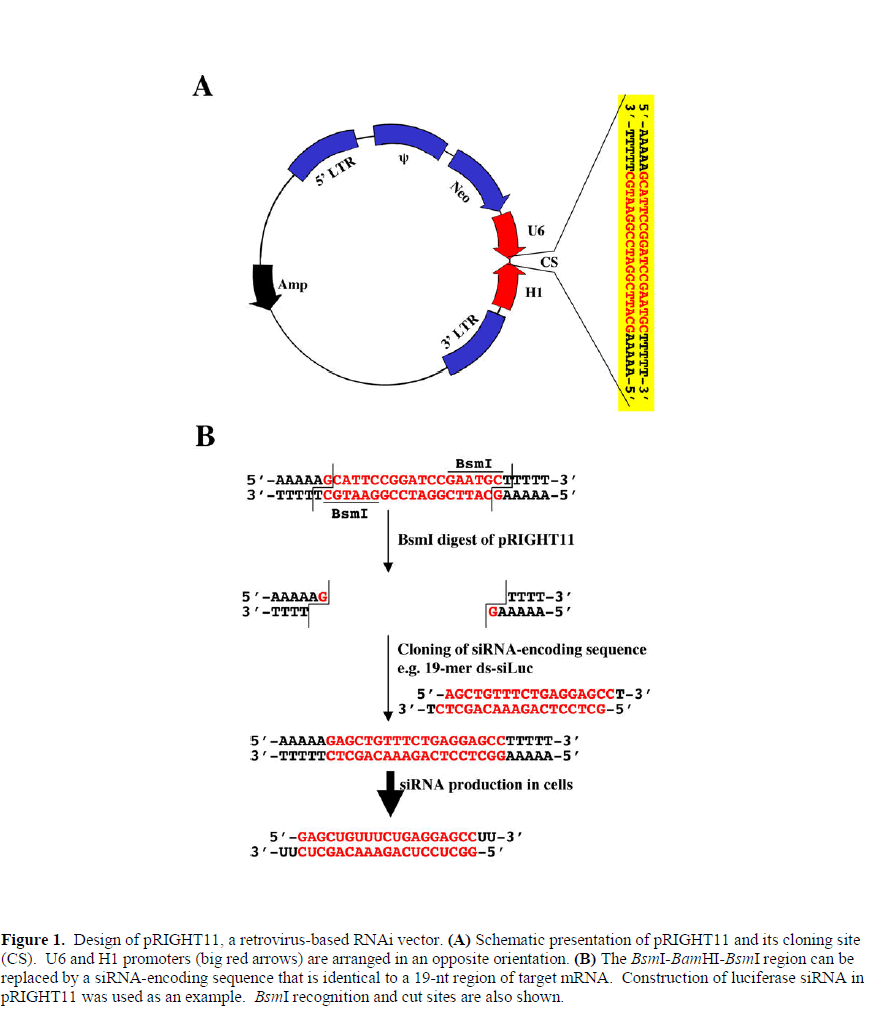
| To construct a retroviral vector for potential genome-wide genetic screen two opposing RNA polymerase III promoters, i.e., U6 and H1, were used. H1 promoter was obtained in a PCR using the following primers: aaggtcgacccatggaattcgaacgctga (forward) and taggatccgaatgctttttagagtggtctcatacagaac (reverse). U6 promoter was amplified from total human genomic DNA by PCR using the following primers: aataagcttaacgcgtagtggaaagacgcgcaggca (forward) and ttcggatccggaatgctttttttcgtcctttccacaag (reverse). The H1 PCR product was digested with SalI and BamHI, and the U6 PCR product was digested with HindIII and BamHI. The two digested DNA were ligated into the HindIII-SalI sites of pLNCX3 (unpublished), a derivative of retroviral vector pLNCX2 (Clontech). The resulting plasmid is named as pRIGHT11 (RNA Interference Gene Hacking/Targeting vector) with BsmI-BamHI-BsmI sites between the two promoters. The restriction enzyme BsmI recognizes a nonplalindromic sequence of GAATGC(1/-1), and its use keeps the mismatch sequences in the 3’ ends of both promoters to only 5 bases (i.e. the AAAAA sequence). A schematic diagram is shown in Figure 1. |
| To construct siRNA-harboring pRIGHT11, siRNAencoding oligonucleotides were synthesized and cloned into the BsmI sites (complete digestion of both sites) of pRIGHT11, creating an expression cassette that theoretically produces both sense and anti-sense 21-22 nt RNA in cells. |
| For making siRNA against luciferase (siLuc: GAGCUGUUUCUGAGGAGCC), the following two oligonucleotides were synthesised: AGCTGTTTCTGAGGAGCCt (sense) and GCTCCTCAGAAACAGCTCt (antisense), and the annealed oligonucleotides were cloned into the BsmIdigested pRIGHT11. Similar cloning strategies were used to generate pRIGHT-siGFP (with a siRNA sequence of GAACGGCAUCAAGGUGAAC, Arteaga et al, 2003), pRIGHT-sip53 (with a siRNA sequence of GACUCCAGUGGUAAUCUAC, Brummelkamp et al, 2002), pRIGHTsiSmad2 (with a siRNA sequence of GGAUGAAGUAUGUGUAAAC) and pRIGHT-siSmad3 (with a siRNA sequence of GGCCAUCACCACGCAGAAC). |
| Cell culture, transfection, luciferase and GFP reporter assays |
| Cell culture, transfection and functional assays were carried out as described (Feng et al, 2002; Lin et al, 2003). Briefly, HeLa and 293T cells were maintained in DMEM supplemented with 10% (v/v) fetal bovine serum. Cells were incubated at 37oC in a 5% (v/v) CO2 incubator. Exponentially growing cells at 30-50% confluency were transfected using LipofectAMINE following the manufacturer’s instructions (Invitrogen). Forty eight hours after transfection, cells were subjected to reporter assays or western blotting. |
| For luciferase assay, plasmid pGL2, which contains the luciferase gene under control of the SV40 early gene promoter (Promega), was used to measure luciferase production. Exponentially grown HeLa cells at 30% confluency were transfected with pGL2, pSVJgal (J- galactosidase expression plasmid as an internal control), and pRIGHT-siLuc or pRIGHT11 control vector. The amount of transfected DNA was 0.5 Kg per well (12-well plate). The ratio of pGL2:pSVJgal:pRIGHT was 2:1:5. Cells were then harvested for measurement of luciferase and J-galactosidase activities. All assays were done in duplicate and repeated more than 3 times, and all values were normalized for transfection efficiency against J- galactosidase activity. |
| For GFP assay, plasmid pEGFP, which contains the enhanced GFP under control of CMV promoter (Clontech), was transfected, together with/without pRIGHT-siGFP, into HeLa cells. Forty eight hours after transfection, production EGFP was first examined by assessing production of green fluorescence under Zeiss Axioplan II microscope, and then by western blotting. In western blotting, EGFP was detected with anti-GFP antibody (Santa Cruz Biotech) and visualized by chemiluminescence (Pierce). |
| Results And Discussion |
| Design of pRIGHT11 – an RNAi-based vector for genetic screens |
| RNA Polymerase III promoters U6 and H1 have been used to drive expression of siRNA (Brummelkamp et al, 2002; Paddison et al, 2002; Sui et al, 2002). Currently there are two ways to produce siRNA in cells. One is to design a stem-loop hairpin cassette that contain the following structure: N19(sense strand)-Loop-N19(antisense strand) (Brummelkamp et al, 2002; Paddison et al, 2002; Sui et al, 2002). The RNA product will contain a base-paired stem, which can be processed by cellular Dicer to become double-stranded ~21-mer siRNA. The other way is to express the N19 sense and the antisense in a different cassette (which can be on the same or different vector) (Miyagishi and Taira, 2002). The individually expressed sense N19 and anti-sense N19 will match by base pairing in cells. However, either expression vector cannot be used as a vector for efficient library carrier. Mathematically, a random library would be enormously large (419+19 = 438 = ~7.6x1022), impossible for genetic screen. |
| By taking advantage the fact that nucleotide sequences close to the initiation site of U6 or H1 are not critical for promoter specificity and efficiency, we designed a vector in such a way that both promoters will be oriented head-tohead, so that one promoter will drive the expression of the sense strand and the other expresses the antisense strand (Figure 1). In addition, the U6 promoter preferably initiates transcription from a G and the H1 promoter also prefers G or A, we can further reduce the size of library by using G as the first 5’ nucleotide on both strands. The resulting random sequence will be G(N17)C. Figure 1 shows schematic presentation of the vector design. |
Proof of principle using reporter assays |
| To provide proof the principle of our vector design, we chose two common reporter genes, i.e., luciferase and EGFP. The siRNA target sequences for these two reporter genes are described in the “Materials and Methods”. By cloning corresponding DNA oligonucleotides (that encode siRNA) into our vector to replace the G(N17)C region, we created pRIGHT-siLuc and pRIGHT-siGFP. |
| We next transfected 293T cells with pGL2 luciferase expression vector with or without pRIGHT-siLuc, and then measured the luciferase activity in transfected cells. We found that pRIGHT-siLuc can effectively knock down the expression of the luciferase production, achieving a 80% reduction in comparison to the pRIGHT11 vector control (Figure 2A). In contrast, pRIGHT-siGFP and pRIGHT11 vector had no effect. The effectiveness of the pRIGHT-produced siRNA was comparable to that of siRNA/shRNA produced by H1-carrying pSUPER vector. In a similar experiment where we introduced GFP expression vector together with pRIGHT-siGFP, we found that pRIGHT-siGFP can suppress the expression of GFP protein. As shown in Figure 2B, western blotting analysis showed a dramatic reduction of the GFP level in the presence, but not in the absence, of pRIGHT-siGFP. Furthermore, fluorescence microscopy also indicated the suppression of green fluorescence production by pRIGHTsiGFP, but not by the control vector (Figure 2C). |
 |
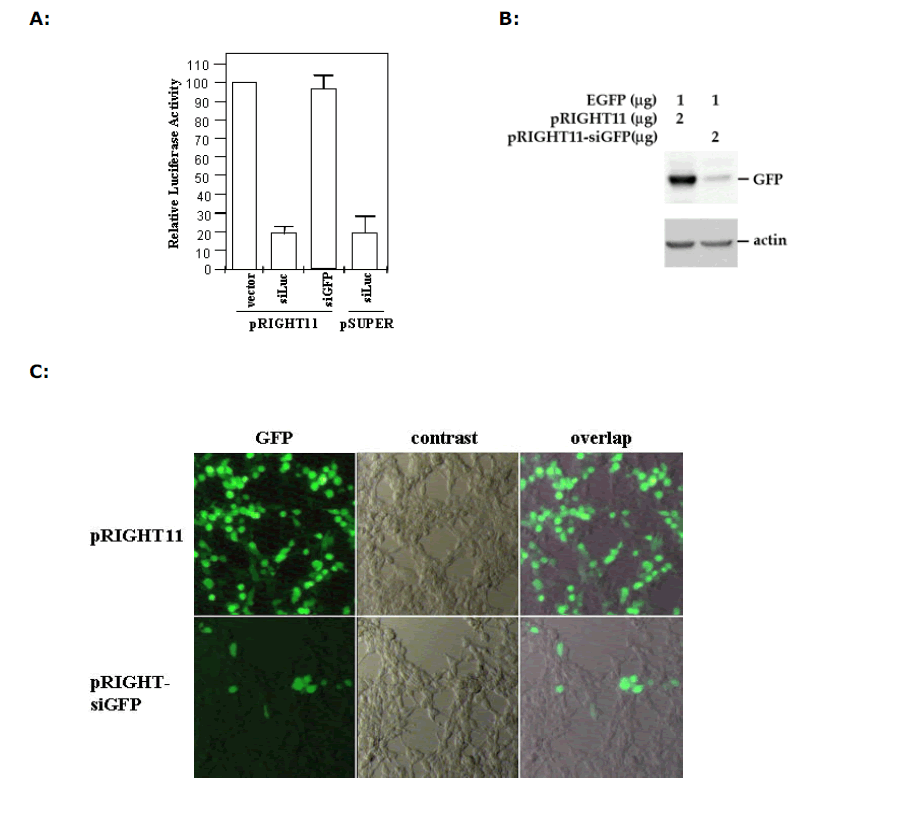 |
| Figure 2. Sequence-specific inhibition of reporter gene expression. (A) Chemilumiscent analysis of luciferase expression. HeLa cells were transfected with luciferase-expressing vector pGL2 together with/without pRIGHT-siLuc. Luciferase expression was determined by chemiluminescence. Luciferase activity in the empty vector-transfected cells is considered as 100%. siLuc and siGFP express siRNA against luciferase and GFP, respectively. pRIGHT-siLuc and pSUPER-siLuc have similar effects in suppression of luciferase expression. (B) Western blotting analysis of GFP expression. HeLa cells were transfected with pEGFP and the GFP protein was analyzed by SDS-PAGE, followed by western blotting using an anti-GFP antibody. pRIGHT-siGFP expressed siRNA against GFP. pRIGHT11 is an empty vector. (C) Microscopic analysis of GFP expression in HeLa cells. GFP was visualized by fluorescent light after transfection with pEGFP in the presence of pRIGHT-siGFP or pRIGHT11 vector. |
| Silencing of natural gene expression |
| We also tested the usefulness of pRIGHT11-produced siRNAs against a natural gene. For this purpose, we constructed pRIGHT-sip53 that expressed p53 siRNA. We observed a robust expression of His-tagged p53 in 293T cells after transfection with an expression for His-p53. Notably, co-transfection of pRIGHT-sip53 suppressed the expression of p53 protein (Figure 3A). |
| To show that pRIGHT11 vector system can be used for knocking down the expression of endogenous genes in mammalian cells, we generated pRIGHT-siSmad2 that expressed Smad2 siRNA. We then introduced pRIGHTsiSmad2 or pRIGHT11 vector into 293T cells (with ~70% transfection efficiency). Results show that endogenous Smad2 expression could be significantly reduced by increased pRIGHT-siSmad2, but not by vector or pRIGHT-siSmad3 (Figure 3B). In contrast, the level of actin remained unchanged. The results of this experiment suggest that pRIGHT11 vector can be used for silencing of endogenous genes in mammalian cells. |
Conclusions |
| In summary, we have designed an expression cassette that expresses both sense and antisense strands of siRNA with opposing polymerase III promoters in mammalian cells. |
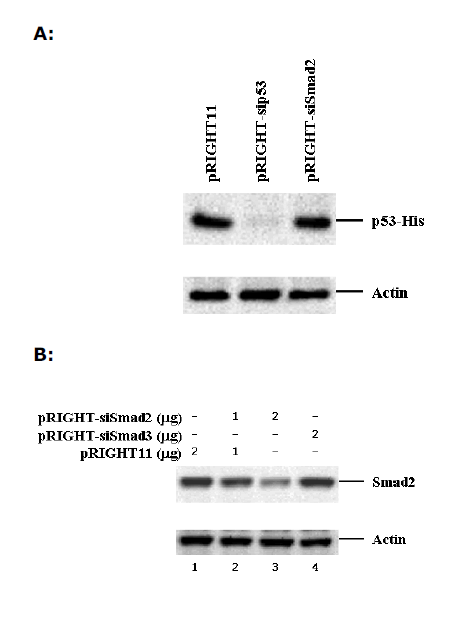 |
| Figure 3. Sequence-specific inhibition of expression of natural genes. A. Inhibition of expression of transfected p53 in 293T cells. 293T cells were transfected with p53-His and the p53 protein was analyzed by SDS-PAGE, followed by western blotting using an anti-His antibody. pRIGHT-sip53 expressed siRNA against p53. pRIGHT-siSmad2 is a non-specific control. B. Inhibition of expression of the endogenous Smad2 in HeLa cells. 293T cells were transfected with pRIGHT-siSmad2 and the Smad2 protein was analyzed by western blotting using an anti- Smad2 antibody. pRIGHT-siSmad2 and pRIGHT-siSmad3 expressed siRNAs against Smad2 and Smad3, respectively. |
| Unlike previously described polymerase III-driven siRNA expression vectors, pRIGHT11 offers the following advantages: |
| 1. Economy: The length of DNA oligonucleotides that are required for cloning siRNA-encoding DNA is reduced by more than half. It significantly reduces the cost of synthesizing DNA oligonucleotides for construction of siRNA expression vector (compared to traditional H1 or U6 vector). For making shRNA in a single polymerase promoter, DNA sequences of the siRNA targets are arranged as “sense-loop-antisense”. For example, two 64- base-long DNA oligonucleotides are needed for making a siRNA-encoding sequence into pSUPER (Brummelkamp et al, 2002). In pRIGHT11, the final length of DNA to be cloned equals to the length of the siRNA target (i.e. 19-23 bp). Yet, pRIGHT11-mediated gene silencing is as effective as pSUPER. |
| 2. Simplicity: The use of a single enzyme BsmI not only further reduced the cost by reducing length of DNA oligonucleotides required for cloning but also makes cloning easier. The annealed siRNA-encoding DNA oligonucleotides can be cloned into pRIGHT11 in either direction. |
| 3. Versatility: pRIGHT11 can be used as a vector for knocking down expression of known individual genes or unknown genes in a genetic screen. We have proven that the pRIGHT vector is useful in knocking down expression of transfected reporter genes such as luciferase and GFP, and also natural genes such as Smad2 and p53. pRIGHT11 can also be used to create stable cell line that express the siRNA of interest by selection of neomycinresistant cells. Most importantly, pRIGHT11 is a retroviral vector and thus, with high infection efficiency retroviruses in mammalian cells, introduction of pRIGHT11 siRNA library into mammalian cells can be used to phenotypic screen in a genome-wide manner. Obviously, the opposing promoter design can be extended to a lentiviral vector that is capable of more efficiently infecting primary cells ( Rubinson et al, 2003; Scherr et al, 2003; Stewart et al, 2003). During the course of our manuscript preparation and review, two studies have used similar opposing promoter in a plasmid-based system (Kaykas and Moon, 2004; Tran et al, 2003). As a retroviral-based vector, pRIGHT11 has greater potential in its application in genetic screens. We have created a retroviral library of GN17C (mathematically a size of 1.7x1010 siRNA molecules) in pRIGHT11. We are currently conducting genome-wide screens for genes involved in TGF-J signaling, cell death, and tumor cell invasiveness. We anticipate that such screens will identify novel genes involved in the targeted cellular responses. |
Acknowledgements |
| This research was supported by the National Institutes of Health (R21CA11293 to X.L., R01GM63773 and R01CA108454 to X.-H.F.). X.-H.F is a Leukemia and Lymphoma Society Scholar. |
Statement Of Competing Interests |
| The authors declared no competing interests. |
List Of Abbreviations |
| TGF-J: transforming growth factor-beta |
References
|