Research Article - Biomedical Research (2017) Volume 28, Issue 8
Computational methods for designing potential inhibitors for activin type IIb (ActRIIB) receptor for treatment of anaemia
Jie Gao1,2, Peng Zhang3, Xiao-qin Zhang2, Xue-feng Zhang2 and Guang-yao Wu1*1Department of Radiology, Zhongnan hospital of Wuhan University, Wuhan, 430071, Hubei province, PR China
2Medical Imaging Department, Inner Mongolia People's Hospital, Hohhot, 010020, PR China
3Department of Imaging Diagnosis, the Affiliated Hospital of Inner Mongolia Medical University, Hohhot, 010010, PR China
- *Corresponding Author:
- Guang-yao Wu
Department of Radiology
Zhongnan hospital of Wuhan University, PR China
Accepted on September 08, 2016
Abstract
Anaemia is a clinical syndrome of blood characterized by decrease in the haemoglobin content in the red blood cells resulting in the marked reduction of the oxygen carrying capacity of the blood. Activins are one of the important types of the Transforming Growth Factor (TGF) protein superfamily. There are various cellular processes in vertebrates from fertilization to adulthood is regulated by the TGF-β superfamily. The heteromeric complexes consisting of type I and type II receptors belonging to the TGFsuperfamily are involved in various signalling processes. The Activin receptor type IIB (ActRIIB) is a transmembrane receptor involved in the negative regulation of red blood cells and skeletal muscle cells. Thus the inhibition of ActRIIB receptor protein results in increased growth of red blood cells and helps to recover anaemic conditions in healthy volunteers as well as in caner or anaemic patients. Molecular docking simulation based in-silico virtual screening technique is used in current experimental study for developing potent inhibitor molecules for activin receptor type IIB of humans for treatment of anaemia. Two compounds ZINC05386901 and ZINC18157167 shows promising results with potent inhibition of the Activin type-II receptor protein of human as well as good pharmacokinetic properties are observed without presence of any toxic effects.
Keywords
Anemia, Activin receptor, Docking, Autodock, Drug.
Introduction
Anaemia is defined as a group of blood disease of humans characterized by either decrease in the count of red blood cells or decreased content of haemoglobin in the red blood cells, resulting in the diminished oxygen-carrying capacity of the blood. The World Health Organisation (WHO) has defined anaemia as the haemoglobin concentration less than 13 g/dl in men and 12 g/dl in women is considered as anaemic condition. The acute anaemia can cause light headedness, tachycardia, dyspnea etc., while the chronic anaemic conditions in human can cause increased risk of cardiac complications, decreased survival, reduced quality-of-life, surgical complications and neurologic dysfunctioning. The common symptoms observed in chronic anaemic conditions are weakness, fatigue, headache, vertigo, faintness, and pallor. Anaemia is classified into different types on the basis of various disease conditions, such as pernicious anemia, aplastic anemia, iron-deficiency anemia and hemolytic anemia [1].
The inadequate productions of red blood cells, increased rate of destruction for red blood cells or blood loss are the major causes for the anaemic conditions. The systemic disorders such as chronic renal disease, malignancy or infectious conditions are manifested through anaemic conditions, thus faster diagnosis of the underlying cause is required essentially. The serum iron level is decreased considerably and may have concurrent folic acid or vitamin B12 deficiency. Anaemia treatment is aimed to correct the pathophysiology involved besides it. Anaemia can affect the people of all age groups but it is common clinical problems mainly observed in the elderly individuals and it is associated with the increased risk of hospitalization and mortality, reduced quality of life and decreased physical functioning. Anaemic conditions are universally common in the critically ill patients and it causes complications in them. There are different types of anaemia out of which some are very rare or mild whereas some types of anaemia are very severe and life threatening if not treated properly [2,3].
The anaemia may either be acquired or inherited. Acquired anaemia is means that the diseased state is developed during the lifetime of the patient and he is not born with it, while inherited anaemia is defined as the gene responsible for the diseased condition is inherited from the parents to the patient.
Anaemia is caused because of various diseases, conditions or factors, such as heavy internal or external bleeding because of injury may cause anaemia or unable to meet increased demand of red blood cells in pregnancy conditions may cause anaemia [3]. The common symptoms observed during anaemic conditions are tiredness, weakness, pale or yellowish skin, faintness or dizziness, increased thirst, increased rate of sweating, weak and rapid pulse, rapid rate of breathing, cramps of lower leg, abnormal heart rhythms etc. The anaemic condition is diagnosed in the patients by both physical examination and blood tests. Physical examination of the colour of skin, gums, and nail beds is performed to identify the anaemic condition in the patient. Complete blood count (CBC) is performed to confirm the presence of anaemic condition in the patient [3].
According to the reports released by the World Health Organisation (WHO) about 66% to 80% of world's total population, i.e. about five billion people are suffering from deficiency of iron and about 30% of world's total population, i.e. two billion peoples are suffering from anaemia [4]. Activins are classified as members of the TGF beta superfamily of signaling molecules and both activin and TGF beta ligands signal through structurally and functionally related serine/threonine kinase receptors haveing various effects on many physiological processes, including cell proliferation, cell death, metabolism, homeostasis, differentiation, immune responses endocrine function, etc. There are three activins (A, B, and AB) that are homo/ heterodimers of two closely related P subunits (PAPA, PBPB, and PAPB).’ Erythroid differentiation factor (EDF) was first found in the culture fluid of phorbol myristate acetate (PMA)-treated human monocytic leukemic cells (THP-1) based on its ability to induce erythroid differentiation of murine Friend erythroleukemia cells. Activins interact with two structurally related serine/threonine kinase receptors, type I and type II, and initiate downstream signaling via Smads to regulate gene expression. Defects in these signaling pathways have been associated with the initiation and progression of the cancer, erythropoisis, and deregulation of muscle mass [5].
Activins induces the synthesis of haemoglobin in humans by enhancing the growth of normal erythroid precursor cells. ActR-IA and ActR-IIB are transmembrane proteins with an extracellular ligand-binding domain and an intracellular kinase domain structurally related to several known serinehreonine kinase present in normal and malignant human myeloid cells possess specific binding sites for activin on their surface. Activins are the enzymes belonging to the transforming growth factor-beta superfamily and are involved in diverse physiological and developmental processes. The antibodies to αvβ3 and αvβ3 integrins block the adhesion of sickle cell. The inhibition of Activin receptor type II protein in humans can increases the red blood cells in healthy volunteers as well as in caner or anaemic patients [5,6]. Here in this present experimental study we have used molecular docking simulation based in-silico virtual screening technique to identify potential inhibitors of Activin Type IIb (ActRIIB) Receptor for Treatment of Anaemia.
Material and Methods
Selection and preparation of protein
The human Activin receptor type II protein bound with its ligand adenine (pdb id-2QLU) was downloaded from RCSB protein data bank shown in Figure 1. The receptor protein Activin receptor type II was prepared for molecular docking by removing ligand from active site, removal of unnecessary water molecules and addition of polar hydrogens. Amino acids ALA266 and HIS268 are present in the active binding site of protein 2QLU.

Figure 1. Crystal structure of the Activin receptor type II of homo-sapiens obtained from RCSB protein data bank. (PDB ID-2QLU)
Preparation of ligand for molecular docking
The substrate ligand cyclic adenine is prepared for molecular docking simulation by providing the rotatable, non-rotatable and un-rotatable bonds present in the ligand.
Identification of binding site
The ligand binding site present in the human Activin receptor type II is identified by using protein visualization softwares like DS Visualizer, Pymol and Chimera. The substrate ligand adenine binds in the receptor’s active binding site as shown in Figure 2.

Figure 2. Bound ligand adenine separated from human Activin receptor type II by using software chimera.
Grid-box formation
Table 1 enumerates the grid parameter points of the grid box used for the molecular docking simulation of human Activin receptor type II protein. These grid parameters were utilized for all docking runs. The grid box is placed in the centre of the ligand by covering all the binding residues involved in the binding of the ligand to ensure that all the extended conformations of ligand fits within the grid box.
| Proteins | x-D | y-D | z-D | Spacing (Ả) | x center | y center | z center |
|---|---|---|---|---|---|---|---|
| 2 QLU | 44 | 42 | 44 | 0.314 | 34.343 | 39.169 | 16.407 |
Table 1. The coordinates of grid box for the three proteins.
Grid maps preparation
The map files for different atom types in ligands and receptor viz. A, OA,C, HD, F, I, Br, Cl, N, NA, SA, S etc. are prepared by running Autogrid utility of the AutoDock suite. These map files prepared by Autogrid is used by AutoDock for carrying out molecular docking simulations.
Docking parameters
Lamarckian genetic algorithm (LGA) is the primary conformational search approach used in AutoDock for molecular docking simulation. A trail population is created for various possible conformations, and these conformations mutate, exchange conformational parameters, and compete in a manner analogous to biological evolution in successive generations for ultimately selecting individuals with lowest binding energy. The individual conformational search for their local conformational space, finding local minima, and then pass this information to later generations is performed by ‘‘Lamarckian’’ aspect, which is its additional feature. The binding free energy of small molecules to macromolecular targets is predicted by using semi empirical free energy force field. The force field allows the incorporation of the intramolecular energies into the predicted free binding energy by evaluating energies for both the bound and unbound states based on a comprehensive thermodynamic model. Docking parameter file for each ligand was prepared by using the 150 Genetic Algorithm (GA) runs, 250000 maximum numbers of evaluations, 27000 maximum numbers of generations and 0.02% rate of gene mutation [7-19].
Docking method validation
The position and orientations of the ligand obtained after the molecular docking study represents potential binding modes of the inhibitors. The various docking parameters considered in the docking methods were validated by redocking individually crystallized ligand adenine over Activin receptor type II of homosapiens. The molecular docking simulation technique is validated by using following parameters:
Binding energy: To validate the molecular docking method the Binding energy of the docked ligand should be in the range between -5 to -15 Kcal/Mol.
Overlay methods: The molecular docking method is validated when the docked conformation of ligand should be perfectly overlaid with the crystal structure of the ligand present in the downloaded protein shown in Figure 1.
Chemical resemblance: The molecular docking method is validated when the docked ligand should have same interactions with the residues of macromolecule as that present in the downloaded crystallized macromolecule.
Selection of chemical libraries
1880 diverse molecules present in the “NCI Diversity Set II” molecular library are virtually screened to identify possible lead compounds. All the ligands used in virtual screening follows Lipinsky’s rule of five for good pharmacokinetic properties. According to Lippinski's rule of five all the ligands should have the following physicochemical parameters in the given range, i.e. ClogP <5, H bond donor <5, H bond acceptor <10, Molecular weight <500.
Virtual screening process
The files necessary for performing virtual screening process were prepared by using software Raccoon. Raccoon is a graphical user interface for AutoDock based virtual screening of ligand libraries against a macromolecule. Raccoon is used to split molecular ligand library file having multiple number of ligands, converts them into the pdbqt format required by AutoDock, and filter the unwanted ligands by using some common criteria, such as Lipinski's rules, fragment-like “rule of 3” and drug-likeness. The input filename, coherent format of grid maps, the presence of non-standard atom types and ensuring that parameters are evaluated at every step for the validation of the process [19-24].
Result analysis of docking simulation
After the screening was over, a script summarize_results.py from Scripps Research Institute was utilized to sort the binding energies of the docked ligand and select the best hits. All the results obtained by molecular docking simulation were evaluated on the basis of hydrophobic and polar interactions obtained between ligand and the binding residues present in the active ligand binding site of the macromolecule and the empirical range of the free binding energy should be in the range of -5 to -15 Kcal/Mol. Binding affinity is calculated from the formula:
Ki=e[(ΔG/RT)]
Where, ΔG=change in free energy upon binding, R=gas constant and T=temperature
Prediction of ADME and toxicity of lead compounds
The toxicity of the virtually screened lead molecules is evaluated by using the OSIRIS online program for toxicity and ADME properties prediction. This program checks the lead molecules for major toxicities such as mutagenicity, tumorigenicity, irritant effect and reproductive effects present in the lead molecule on the basis of functional group present in its chemical structure. This program also calculates drug-likeness and drug score of the lead molecule on the basis of its physicochemical properties.
Results and Discussion
Selection and preparation of protein
The 2QLU protein complex downloaded from RSCB protein data bank consists of a single polypeptide chain of 314 amino acids and a bound ligand cyclic adenine. 141 water molecules present in the receptor molecule are removed from it by using auto dock tools and the receptor is saved in *.pdbqt format.
Preparation of ligand for molecular docking
No rotatable and non-rotatable bonds are present in the ligand molecule adenine. All the bonds of the ligand molecule are unrotatable in nature. The prepared ligand is saved in the *.pdbqt format.
Identification of binding site
Amino acids PHE267, LEU245, THR265, ALA215, LEU328, VAL204, ALA197, LYS196, GLY271, ALA266 and HIS268 are identified as the binding residues present in the active ligand binding site of the Activin receptor type II protein of human.
Grid-box
The coordinates used for the preparation of the grid box are tabulated in Table 1 and 3D pattern is shown in Figure 3.

Figure 3. Three dimensional grid box covering the active ligand binding sites present in the receptor molecule as well as all the residues involved in the binding of the ligand.
Molecular docking simulation
The results obtained after molecular docking of the endogenous substrate ligand adenine (ADE) with the Activin receptor type II of homo-sapiens are presented in Table 2.
| Proteins | Interacting residues | Internal validation rmsd value | Binding energy (Kcal/Mol) | Binding affinity (µM) |
|---|---|---|---|---|
| 4WXQ | PHE267, ALA266 and HIS268 | 0.84 | -5.02 | 132.17 |
Table 2. Molecular docking results of ligand Adenine (ADE) with the Activin receptor type II protein (4O1Z).
Validation of the molecular docking method
The molecular docking process for docking of particular ligand with a specific macromolecule is done by using the following parameters:-
Binding energy: The molecular docking simulation method is validated as the binding energy of the endogenous substrate ligand adenine (ADE) with the Activin receptor type II of homo-sapiens is -5.02 Kcal/Mol, which lies in the predefined range of -5 to -15 kcal/mol.
Overlay methods: The molecular docking method is validated as the docked conformation of the ligand should be perfectly overlaid with the crystal structure of the ligand present in the downloaded protein. The overlaid conformation of the docked ligand with reference to the crystal structure of the downloaded ligand is shown in Figure 4.

Figure 4. The superimposition of the docked conformation of the ligand with reference to its bioactive conformation of the ligand obtained from the crystal structure of the downloaded protein.
Chemical resemblance: The molecular docking method is validated when the docked ligand should have same interactions with the residues of macromolecule as that present in the downloaded crystallized macromolecule. The interactions present in crystal structure are shown in Figure 5A and the interactions present in the docked structure are shown in Figure 5B.

Figure 5. Binding mode and chemical interactions of the bound ligand adenine within the active ligand binding site of human activin receptor type II protein.
Virtual screening
The potential ligands were selected by analyzing the ligand protein interactions for top ranking pose of each ligand and interactions of docked compound were visualized. The molecular docking results of top five molecules selected in Autodock based virtual screening of the Activin receptor type II protein of human with "NCI Diversity set-II" ligand library having 1880 small ligand molecules are shown in Table 3.
| S. No. | Compound ID | Chemical Structure | Binding Energy (Kcal/Mol) |
|---|---|---|---|
| 1 | ZINC05386901 | 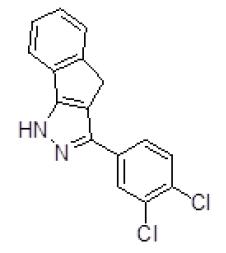 |
-9.66 |
| 2 | ZINC18157167 | 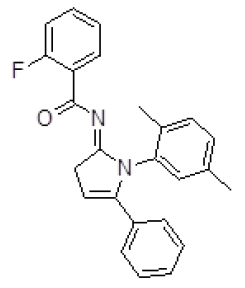 |
-9.45 |
| 3 | ZINC05536814 | 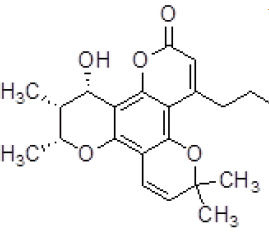 |
-9.41 |
| 4 | ZINC17465958 | 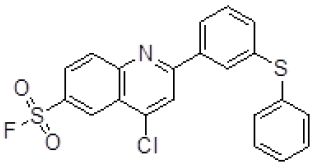 |
-9.13 |
| 5 | ZINC04376856 | 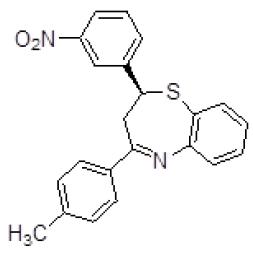 |
-9.12 |
Table 3. Binding energy and affinity of top five hits from "NCI Diversity Set II” after virtual screening against the Activin receptor type II protein of human.
Physicochemical properties evaluation
The best five lead molecules obtained after performing molecular docking simulation based virtual screening of "NCI Diversity set-II" ligand library having 1880 diverse ligand molecules are evaluated for pharmacokinetic profiling by considering important physicochemical properties such as calculated partition coefficient (cLogP), two dimensional Polar surface area (2D PSA), molecular weight, hydrogen bond donor (HBD) and hydrogen bond acceptor (HBA) sites etc. by using online program Osiris Molecular Property Explorer, and Marvin Sketch software. The physico-chemical properties of the five best lead molecules for 5α-reductase enzyme protein of human are shown in Table 4.
| S. No. | Compound ID | Mol. Wt. | ClogP | 2D PSA (Å2) | HBA | HBD |
|---|---|---|---|---|---|---|
| 1 | ZINC05386901 | 260 | 4.1 | 28.68 | 1 | 1 |
| 2 | ZINC18157167 | 383 | 6.06 | 20.31 | 3 | 0 |
| 3 | ZINC05536814 | 370 | 3.95 | 64.99 | 4 | 1 |
| 4 | ZINC17465958 | 429 | 623 | 80.71 | 3 | 0 |
| 5 | ZINC04376856 | 374 | 5.25 | 65.73 | 4 | 2 |
Table 4. “Lipnski’s rule of five” for the hits on the Activin receptor type II protein protein.
ADME and toxicity profiling of lead compounds
The pharmacokinetic parameters such as absorption, distribution, metabolism, excretion and toxicity were evaluated by using online server Osiris Molecular Property Explorer and following results were obtained. All the five best virtually screened lead molecules are tested for their pharmacokinetic parameters and presence of any major toxic effect and it is observed that two out of five selected lead molecules, i.e. ZINC05386901 and ZINC18157167 are having good pharmacokinetic profile with the presence of no or very low toxic effects. All the five selected lead molecules are also evaluated for various parameters like drug-likeness and Drug Score values and found that the two molecules ZINC05386901 and ZINC18157167 are having these parameters in acceptable range. Whereas remaining lead molecule shows poor pharmacokinetic profile with presence of some major toxic effects like mutagenicity, tumorogenicity and some reproductive effects. The ADME and toxicity results of best lead molecules obtained after virtual screening are shown in Figures 6-10.

Figure 6. ADME Profiling and toxicity prediction of ZINC05386901 molecule by using online Molecular Properties Prediction Server- Osiris Property Explorer.

Figure 7. ADME Profiling and toxicity prediction of ZINC18157167 molecule by using online Molecular Properties Prediction Server- Osiris Property Explorer [18].

Figure 8. ADME Profiling and toxicity prediction of ZINC05536814 molecule by using online Molecular Properties Prediction Server- Osiris Property Explorer [18].

Figure 9. ADME Profiling and toxicity prediction of ZINC17465958 molecule by using online Molecular Properties Prediction Server- Osiris Property Explorer [18].

Figure 10. ADME Profiling and toxicity prediction of ZINC04376856 molecule by using online Molecular Properties Prediction Server- Osiris Property Explorer.
Conclusion
Molecular docking simulation based in-silico virtual screening using Autodock4 was very useful in short-listing potential lead molecules. Two compounds ZINC05386901 and ZINC18157167 shows promising results with potent inhibition of the Activin type-II receptor protein of human as well as good pharmacokinetic properties are observed without presence of any toxic effects. These molecules serves as promising lead molecules for further structure based discovery of novel drugs for treatment of anaemia.
Acknowledgement
This study has been supported by major program of the National Natural Science Foundation of China (Grant No. 81227902 and National key basic research program (Grant No. 2016YFC1304702).
References
- Dipiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey LM. Pharmacotherapy- A Pathophysiologic Approach. 7th Ed, McGraw-Hill, New York, USA.
- Suragani RN, Cadena SM, Cawley SM, Sako D, Mitchell D. Transforming growth factor-Î2 superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat Med 2014; 20: 408-414.
- Patel S, Shah M, Patel J, Kumar N. Iron deficiency anemia in moderate to severely anaemic patients. Guj Med J 2009; 64: 15-18.
- Raghupathy R, Billett HH. Promising therapies in sickle cell disease. Cardiovasc Hematol Disord Drug Targets 2009; 9: 1-8.
- Hildén K, Tuuri T, Erämaa M, Ritvos O. Expression of type II activin receptor genes during differentiation of human K562 cells and cDNA cloning of the human type IIB activin receptor. Blood 1994; 83: 2163-2170.
- Charles CV, Summerlee AJ, Dewey CE. Anemia in Cambodia: prevalence, etiology and research needs. Asia Pac J Clin Nutr 2012; 21: 171-181.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009; 30: 2785-2791.
- Tuccinardi T. Structure-based virtual screening: identification of novel CB2 receptor ligands. Bioorg Med Chem Lett 2007; 4: 15-19.
- von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov 2007; 6: 967-974.
- Phosrithong N, Ungwitayatorn J. Molecular docking study on anticancer activity of plant-derived natural products. Med Chem Res 2009; 19: 817-835.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009; 30: 2785-2791.
- Phoristhong N. Molecular docking study on anticancer activity of plant-derived natural products. Med Chem Res 2009; 19: 817-835.
- Patani GA, LaVoie EJ. Bioisosterism: A Rational Approach in Drug Design. Chem Rev 1996; 96: 3147-3176.
- Hansch C, Garg R, Kurup A. Searching for allosteric effects via QSAR. Bioorg Med Chem 2001; 9: 283-289.
- Menaa F, Braghini CA, Vasconcellos JP, Menaa B, Costa VP. Keeping an eye on myocilin: a complex molecule associated with primary open-angle glaucoma susceptibility. Molecules 2011; 16: 5402-5421.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001; 46: 3-26.
- Wong CF, McCammon AJ. Protein simulation and drug design. Adv Protein Chem 2003; 66: 87-121.
- Gorelik B, Goldblum A. High quality binding modes in docking ligands to proteins. Proteins 2008; 71: 1373-1386.
- http://fruity-updates.blogspot.in/2011/11/protein-docking-aatu-kaapro-janne.html
- Iorga B. Acetylcholine nicotinic receptors: finding the putative binding site of allosteric modulators using the “blind docking” approach. J Mol Mod 2006; 12: 366-372.
- B-Rao C, Subramanian J, Sharma SD. Managing protein flexibility in docking and its applications. Drug Discov Today 2009; 14: 394-400.
- Larsen PK, Liljefors T, Madsen U. Textbook of drug Design and Discovery, 3rd Ed, CRC Press, USA, 2009.
- Mujwar S, Pardasani KR. Prediction of Riboswitch as a Potential Drug Target for Infectious Diseases: An Insilico Case Study of Anthrax. J Med Imag Health Inform 2015; 5: 1-10.
- Anderson RA, Cambray N, Hartley PS, McNeilly AS. Expression and localization of inhibin alpha, inhibin/activin betaA and betaB and the activin type II and inhibin beta-glycan receptors in the developing human testis. Reproduction 2002; 123: 779-788.