- Journal of RNA and Genomics (2012) Research Report
Anti-PML-RARa shRNA sensitises promyelocytic leukaemia cells to all-trans retinoic acid
| Nicholas P Casey* and Gregory M Woods Menzies Research Institute, University of Tasmania, Medical Science 1, 17 Liverpool Street, Hobart TAS 7000, Australia |
| Corresponding Author: Nicholas P Casey, Email: npcasey@utas.edu.au, Tel: +61 3 62264832, Fax: +61 3 62264816 |
| Received 02 February 2012; Revised 10 July 2012; Accepted 11 July 2012; Published 13 July 2012 |
| © Copyright The Author(s): Published by Library Publishing Media. This is an open access article, published under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.5). This license permits non-commercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
Visit for more related articles at Journal of RNA and Genomics
Abstract
The PML-RARα fusion gene disrupts the retinoic acid differentiation signal in a range of leukaemia types, promoting proliferation. We designed a shRNA to target the fusion mRNA, and the shRNA expression cassette was delivered via lentiviral transduction. Delivery of this shRNA significantly down regulated the target mRNA, with effects also evident at the protein level. When combined with ATRA administration, this down regulation of the fusion gene strongly inhibited proliferation in the NB4 PML cell line.
Keywords |
| Leukaemia, PML, shRNA, lentiviral vector |
Introduction |
| Acute promyelocytic leukaemia (APL) forms a subset of acute myeloid leukaemia (AML), identified as AML-M3 according to the French-American-British (FAB) classification and APL with t(15;17)(q22;q12);PML-RARα according to the WHO classification, with the chromosomal translocation t(15:17), involving the retinoic acid receptor alpha (RARα) and the PML gene (reviewed in Parmar and Tallman, 2003). The leukaemogenic effects of these translocations result from a dominant negative inhibition of transcription regulation (Lin et al, 2001). |
| While chimeric forms of the RARα receptor are insensitive to physiological concentrations of retinoic acid, APL can exhibit a dramatic response to higher, pharmacological doses of retinoic acid, in the form of all-trans retinoic acid (ATRA), however remissions with ATRA monotherapy were short lived and some cases are unresponsive altogether (reviewed in Parmar and Tallman, 2003). Combinatorial induction therapy with ATRA and chemotherapy can achieve event free survival of up to 88% at two years, and a range of chemotherapies have been tested in combination with ATRA. This combined approach also results in a lower incidence of retinoic acid syndrome (RAS) compared with ATRA alone. |
| Post-remission maintenance therapy using ATRA with or without chemotherapy is also beneficial (reviewed in Parmar and Tallman, 2003). Nevertheless, of patients initially treated with ATRA plus chemotherapy, 20-30% will eventually relapse. While secondary complete remission after ATRA is common, long term duration of the second remission is rare. In cases of relapse after ATRA, or in cases that are refractory to ATRA treatment, arsenic tetroxide remains an effective option, with complete remission rates of 87% (reviewed in Parmar and Tallman, 2003). Despite these high rates of initial and secondary remission, patients experiencing subsequent relapse are at the greatest risk of early death, and require intensive post-remission therapy, often including autologous of allogeneic stem cell transplantation. When allogeneic transplantation is an option, relapse rates are lower than for autologous transplants (15% and 44% respectively, at six years), however there is an elevated risk of transplant related mortality (33% and 25%, respectively, at six years) (reviewed in Parmar and Tallman, 2003). |
| Due to the risk of relapse or resistance present with all existing conventional therapies, there is ongoing interest in developing molecular therapeutic options. The three main strategies explored to date are hammerhead ribozymes, antisense oligonucleotides, and short-interfering RNA. |
| Hammerhead ribozymes targeted to the fusion mRNA transcript were able to down-regulate PML-RARα mRNA, inhibiting growth and inducing apoptosis, but did not overcome the maturation block (Nason-Burchenal et al, 1998). Antisense oligonucleotides targeted to the same transcript sequence lead to a reduction in levels of the fusion mRNA, inducing partial differentiation and apoptosis (Chen et al, 1999). Finally, a 30nt short hairpin RNA (shRNA) has been shown to reduce levels of the fusion protein, however effects on cell proliferation were not examined (Oshima et al, 2003). |
| These results illustrate the potential of molecular strategies in supporting or replacing existing therapies for cancer, however they do not address the issue of delivery. Key to the efficacy of molecular strategies such as RNAi, is sustained delivery. One of the foremost candidates for the delivery of stable gene therapy are lentiviral vectors, which are capable of stably transducing quiescent cells, and have proven to be particularly efficient in the transduction of hematopoietic stem cell types, including CD34+ lineages (Salmon et al, 2000). The primary aim of this study, therefore, was to test lentiviral transduction for delivery of novel shRNAs designed to target the PML-RARα fusion gene, as a strategy for down regulating this gene and inhibiting proliferation of the APL cells. It was envisaged that such a strategy would complement existing autologous transplantation protocols, and might provide the option of generating a population of stem cells that are ‘pre-treated’ to resist the downstream effects of the PML-RARα translocation, and thus might be suitable for autologous transplantation into patients for whom other treatment options are not available or are ineffective. |
Materials and Methods |
| Hairpin design |
| In order to generate novel shRNA designs, the sequence of the PML-RARα molecular fusion was submitted to shRNA design modules on both the Ambion (http://www.ambion.com/techlib/misc/siRNA_finder.html) and GenScript (https://www.genscript.com/ssl-bin/app/rnai) websites. In addition, a 30nt hairpin design centred upon the breakpoint (Oshima et al, 2003), was adapted to a 21nt hairpin design, for testing alongside the algorithm-derived sequences. Manual screening of the resulting shRNA designs to include those that spanned the fusion breakpoint resulted in only two unique shRNA designs, designated PR1 and PR2. Their alignments to the fusion cDNA were as follows; |
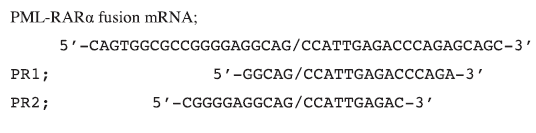 |
| The inverted repeats of the shRNA design were ordered as ssDNA oligonucleotides, annealed by heating and slow cooling, then purified and cloned into the multiple cloning site adjacent to the H1 RNA Polymerase III (Pol III) promoter of the 3.1-H1 neo pSilencer vector (Ambion). The entire expression cassette was amplified by PCR using primers with Xma I linkers, then cloned into the single Xma I site of the pHIV-7-GFP lentivirus vector, which was generously provided by Professor John Rossi (Li et al, 2003). |
| These hairpin designs and cassette orientations were tested in preliminary trials (see Results and Discussion, Figure 1), with the PR1 shRNA design used exclusively thereafter. Further preliminary testing of shRNA cassette orientation relative to other genes within the expression vector indicated that the shRNA expressed from a cassette in the reverse orientation was most effective in down-regulating the target mRNA (results not shown). In order to check for a non-specific response to the PR2 shRNA design, transduced cells were also examined for up regulation of 2’,5’-oligoadenylate synthetase (OAS1) (Bass, 2001). This marker gene was not affected by expression of the PR2 shRNA design (results not shown), and so this design was considered suitable for further study. |
| A control hairpin with an inversion of the 4 central nucleotides was tested against both the PR2 shRNA, and the original nonsense (NS) shRNA of the (Ambion) expression cassette. Despite the 4nt difference, this inverted control was also found to affect the target mRNA, and so was not used. Other candidate control designs (Whither RNAi?, 2003) showed considerable homology with normal human genes, and so the NS shRNA was used as the control throughout this study. |
| Vector production |
| Vector particles were generated by calcium phosphate precipitation (Sambrook and Russell, 2000) 293T cells growing in DMEM (Gibco) with 10 % (v/v) FCS, L-glutamine and antibiotics. The pHIV-7-GFP vector plasmid was co-transfected with the pMDG.2 and psPAX2 accessory plasmids provided by Dr Didier Trono (Tronolab, Lausanne, Switzerland). The culture supernatant was harvested at 48 and 72hr post-transfection, and the virus particles concentrated and pelleted by ultracentrifugation (Geraerts et al, 2005), and resuspended in serum-free RPMI before freezing. |
 |
| NB4 cell culture |
| The NB4 cell line carries the PML-RARα molecular fusion and was isolated from the marrow of an APL patient in relapse (Lanotte et al, 1991). These cells were cultured in RPMI medium (Gibco) with 10 % (v/v) FCS, L-glutamine and antibiotics. The NB4 cells were transduced with purified vector particles in two rounds, in serum-free medium, with 6ng/μl polybrene. |
| Multiplex qRT-PCR |
| Total RNA collected using Trizol (Invitrogen) as per the manufacturer’s instructions, and mRNA levels were analysed using a RotorGene 3000 (Qiagen) with Plexor One-Step qRT-PCR System (Promega) and compatible labelled primers (Biosearch Technologies). The β-2-microglobulin housekeeping gene was used as a control (Schmittgen and Zakrajsek 2000), and the primers sequences were as follows; |
| Forward primer - 5′-TGCTTACATGTCTCGATCCCACTTAAC-3′, labelled with a 5′ CAL Fluor Red 610 fluorophore |
| Reverse primer - 5′-CTGGTCTTTCTATCTCTTGTACTACACTGAAT-3′. |
| The primers used specific to the PML-RARα fusion sequence were as follows; |
| Forward primer - 5′-CTGCTGCTCTGGGTCTCAATG-3′, 5′ labelled with the 6-FAM fluorophore |
| Reverse primer - 5′-CCAGGAGCCCCGTCATAGGAA-3′ |
| For quantitative analysis, Rotorgene output for the PML-RARα fusion mRNA was normalized to the housekeeping gene, and then results from the PR2 treated cells were compared with the NS treated controls by paired t-test. |
| Western blots |
| To obtain nuclear proteins, cells were cooled, and then prepared as follows, at 4°C. Wells were washed with PBS, then resuspended in Buffer A, with Igepal (1mM Tris (pH 7.4), 10mM NaCl, 3mM MgCl2, 25μM EDTA (pH 8.0), 0.2% (v/v) Igepal, in MilliQ water). Following incubation and centrifugation, the supernatant, containing the cytoplasmic fraction, was removed. The nuclear pellet was then resuspended in Buffer A without Igepal (10mM Tris (pH 7.4), 10mM NaCl, 3mM MgCl2, 0.1mM EDTA (pH 8.0), in MilliQ water). Following incubation and centrifugation, the pellet was resuspended in Buffer C (400μl 0.4M NaCl, 7.5mM MgCl, 0.2mM EDTA in MilliQ water, with 1mM dithiorethrol added immediately before use). This was again incubated and centrifuged, and the supernatant, containing the nuclear proteins, was removed and stored at -80°C until use. Proteins were separated by electrophoresis on a 12 % (w/v) polyacrylamide gel as per standard protocols, then blotted onto a PVDF membrane, and stained with the appropriate antibodies. |
| Proliferation assays |
| Cell proliferation assays were performed as per the manufacturer’s instructions, using the colorimetric ‘Cell Titer 96’ cell proliferation assay (Promega) and the Spectra max M2 plate reader, and SoftMax Pro 4.2 software (Molecular Devices, Surrey Hills, Australia). |
| Differentiation assay |
| Cells were incubated for 15min with either CD11b or CD14 antibodies (BD Biosciences, North Ryde NSW, Australia) and then fixed for analysis by flow cytometry on Coulter EPIGS ELITE ESP fluorescence activated cell sorter (FACS) machine. |
Results and Discussion |
| shRNA selection and mRNA knockdown |
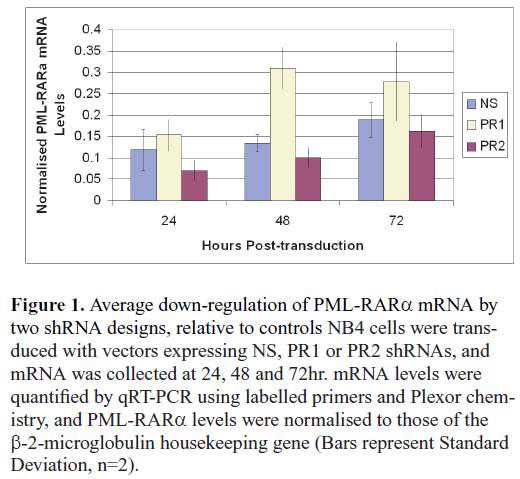
| A preliminary study comparing the activity of the two candidate shRNA designs (PR1 and PR2) against a nonsense (NS) control found that the PR2 design consistently down-regulated levels of the target mRNA (Figure 1). By contrast, the PR1 shRNA appeared to boost levels of the target mRNA. While the reasons for this effect were unclear, further examination was beyond the scope of this study. The PR1 design was discarded. |
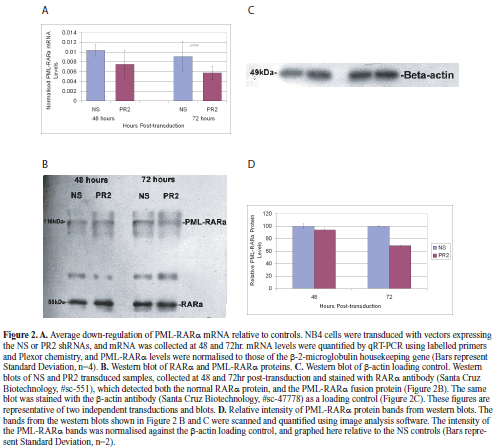
| With the vector construct used in this study, we found that we were able to achieve transduction of 84.8% of cells, as assessed by flow cytometry. Total RNA was collected at 48 and 72hr post-transduction, and normalized values of PML-RARα mRNA copy number were analysed by a pair-wise comparison between the NS and PR2 transduced populations. This analysis demonstrated that the PR2 shRNA design was effective at down regulating the target mRNA (Figure 2A; knockdown to 73% at 48hr, and 63% at 72hr), and that this difference was significant at 72hr (Paired t-test, p<0.05, n=4). Thus it was clear that the PR2 shRNA expressed from this vector was able to effectively down-regulate the fusion mRNA in the period shortly after transduction. |
| To examine the effect of PR2 shRNA transduction on levels of the PML-RARα protein, nuclear extracts were also collected at 48 and 72hr post-transduction. Despite the 24hr half-life of this protein (Grignani et al, 1999), we found evidence that at least a sub-set of the NB4 cells were continuing to proliferate (see below), and so we felt there was little point in examining protein levels beyond 72hr. Nevertheless, we did find evidence for down-regulation of PML-RARα protein levels at 72hr post-transduction, with normalized intensity of the fusion gene band at 68.82% of that found in the control cells (Figures 2B, 2C, 2D). The finding that levels in PR2-transduced cells were approximately equivalent to the controls at 48hr (94%), despite down-regulation of mRNA at this time-point, is consistent with the half-life of this protein. |
| Effects on cell proliferation |
| Down-regulation of the PML-RARα transcript has been shown to inhibit proliferation and induce apoptosis (e.g., Nason-Burchenal et al, 1998; Chen et al, 1999), so we performed cell proliferation assays at 24, 48 and 72hr post-transduction. A marked and significant difference (Paired t-test, p<0.05) in cell number was evident at 24hr post-transduction, with the PR2 cells lagging behind the controls (Figure 3). Trypan blue assays were conducted at this time-point and suggested increased rates of non-viable PR2 cells, however this difference was not significant (data not shown). At 48 and 72hr post-transduction, while the PR2-transduced cells still lagged significantly behind the controls (Figure 3), the margin was greatly reduced, and the PR2 cells were proliferating rapidly. |
 |
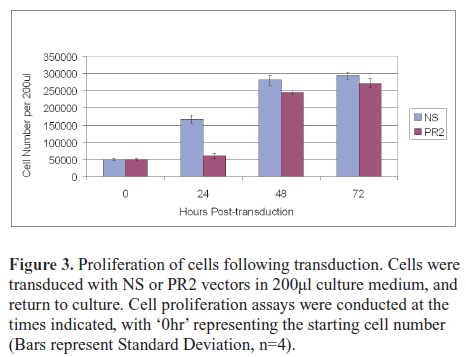 |
| Given that initial rates of GFP marker gene expression in both transduced cell populations were in the order of 85-90 % (data not shown), it was probable that a subpopulation of cells had escaped PR2-transduction and were responsible for the high rate of proliferation observed between 24 and 48hr. |
| Purification |
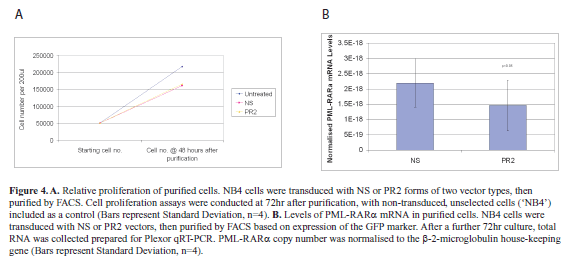
| In order to address this point, cells were transduced with NS or PR2 expressing vectors, then FACS-sorted for marker gene (GFP) expression after 48hr. Cell proliferation assays were again performed after a further 72hr of culture (Figure 4A). From these assays it was clear that purified PR2-transduced cells were able to proliferate at a rate comparable to the NS-transduced controls (Paired t-test, p<0.05, n=4). |
| This result was contrary to expectations following effective down-regulation of the PML-RARα fusion gene. Although the cells had been FACS-purified, and therefore these populations all carried the vector, there is evidence that vector copy number can influence RNAi efficiency (Scherr et al, 2005). We therefore analyzed expression of the GFP marker transgene in transduced cells 48 hours after sorting. Mean fluorescence intensity of the NS-transduced cells (28.4 arbitrary fluorescence units) was greater than in the PR2-transduced cells (16.1 arbitrary fluorescence units). While this assay cannot distinguish between vector copy number and transgene expression levels, the lower fluorescence in the PR2-transduced cells was consistent with selection against high RNAi activity in these cells. |
 |
| Proliferative expansion of a sub-population of PR2-transduced cells with lower shRNA activity. |
| In light of these results, before progressing further we sought to confirm that down-regulation of the fusion mRNA persisted in the purified cell populations. To do this, cells were transduced as above, sorted at 48hr post-transduction, then cultured for a further 72hr after sorting. Total RNA was collected and prepared, and normalized PML-RARα mRNA copy number assessed, as above. This qRT-PCR analysis (Figure 4B) confirmed that despite probable lower levels of PR2 shRNA expression, PML-RARα mRNA was persistently expressed in the purified cells, with significant down-regulation in the PR2-transduced population compared with the controls (64.62% of controls; paired t-test, p<0.05, n=4). |
| From these experiments it was therefore clear that while the PR2 shRNA was able to down regulate the PML-RARα fusion gene, this alone was not sufficient to impair cell proliferation. Consequently, we wanted to test whether the down-regulation of PML-RARα had any effect on mediation of the retinoic acid pro-differentiation signal, the disruption of which is crucial to the oncopathology of the PML-RARα fusion (de The et al, 1989). Furthermore, because of the risks associated with sustained ATRA therapy, we were interested to see whether the PR2 transduced cells responded to the retinoic acid signal at a lower dose of ATRA than the control cells. In order to do this, we identified and used an intermediate dose of ATRA (50ng/ml) that had only a modest effect on proliferation of non-transduced cells (see below) in order to assess whether the PR2 transduced cells exhibited a greater response than the controls. |
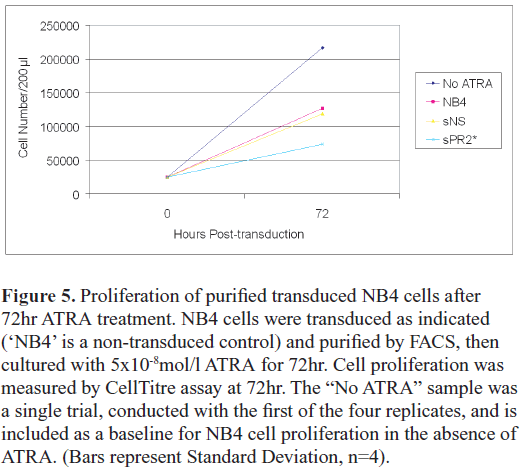
| As retinoic acid signalling inhibits the proliferation of APL cells, and triggers their differentiation, we first examined the effect of ATRA on proliferation of the transduced cells. Cells were transduced, purified after 48hr, then cultured in 50ng/ml ATRA for a further 72hr, at which time the proliferation assays were performed (Figure 5). Non-transduced cells treated with ATRA (‘NB4’) and non-transduced cells cultured without ATRA (‘No ATRA’) were both included as controls. As indicated above, the ATRA had a modest effect on the proliferation on the non-transduced controls. The effect of ATRA on the NS-transduced cells was very similar to the non-transduced control, however proliferation of the PR2-transduced cells was significantly impaired in the presence of ATRA (NS vs PR2, paired t-test, p<0.05, n=4). |
 |
| Myeloid differentiation markers |
| Having established that the PR2shRNA sensitized the NB4 cells to the anti-proliferative effects of ATRA signalling, we examined the effects of this treatment upon differentiation of the cells. After transduction, purification, and culture as described above, cells were cultured in 50ng/ml ATRA for a further 72hr, then labelled with myeloid cell surface markers, CD14 or CD11b. There was little difference in CD11b and CD14 staining (not shown), and so it was concluded that PR2-transduction and ATRA treatment had no meaningful effect on cell differentiation. Thus, while there was no evidence of differentiation, it remains clear that the PR2 shRNA was strongly sensitizing the NB4 cells to the anti-proliferative effects of ATRA signalling. In these respects, our results closely resembled those described using hammerhead ribozymes, where effects on proliferation, but not differentiation, were observed (Nason-Burchenal et al, 1998). |
Conclusions |
| In this study, we have demonstrated as follows: |
| • Despite down-regulation of the PML-RARα fusion gene by a targeted shRNA, a substantial proportion of the transduced cells retained the ability to proliferate |
| • Such knockdown might be considered necessary, but not sufficient, for inhibition of APL cell proliferation. |
| • We further demonstrated that down-regulation of the PML-RARα fusion gene sensitized the APL cells to retinoic acid (ATRA) signalling. |
| • This last finding may lead to additional options for strategies based on ex vivo treatment and retransfusion of autologous stem cells. |
Acknowledgements |
| This work was funded by a David Collins Leukaemia Foundation grant. NPC held a University of Tasmania Strategic Scholarship. |
Statement of Compteting Interests |
| None declared. |
List of Abbreviations |
| APL; Acute Promyelocytic Leukaemia |
| ATRA; All-Trans Retinoic Acid |
| FACS; Fluorescence-Activated Cell Sorting |
| GFP; Green Fluorescent Protein |
| mRNA; messenger Ribonucleic Acid |
| PML; Promyleocytic Leukaemia |
| RARα; Retinoic Acid Receptor α |
| qRT-PCR; quantitative Reverse Transcription Polymerase Chain Reaction |
| shRNA; short hairpin Ribonucleic Acid |
References
|