- Journal of RNA and Genomics (2007) New Methods and Technologies
A strategy for constructing and verifying short hairpin RNA expression vectors
| Fang-Jun Jia1, Mei Huang1, Yuan-Chang Yan2, Yi-Ping Li 1,* 1Laboratory of Molecular Cell Biology, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China; 2Model Organism Division, E-Institutes of Shanghai Universities, Shanghai, China |
| Corresponding Author: Yi-Ping Li, Email: oocyte@sunm.shcnc.ac.cn, Tel: +21 54921415, ext 5951, Fax: +21 54921011 |
| Received 21 October 2006; Revised 01 December 2006, Accepted 15 December 2006; Published online 23 January 2007 |
Abstract
The application of RNA interference (RNAi) to study gene function is now commonplace in a variety of biological systems. Producing short hairpin RNA (shRNA) by DNA vectors is one popular strategy for RNAi applications. Here, we describe a one-step PCR method, termed reverse PCR, for constructing shRNA expression vectors. Characteristically, the pair of primers binds to circular plasmid in a back-to-back manner. The anchored primers provide the templates of shRNA sense strand and antisense strand locating to the two separate ends of PCR segment, which will benefit the PCR amplification and subsequent cloning by avoiding premature formation of a hairpin configuration. Finally, the establishment of a circular vector is achieved by self-ligation of the single PCR product. In addition, our results indicated that the hairpin loop including a single restriction site is resistant to digestion, while inclusion of twin restriction sites in the loop leads to activity, creating an optimal strategy for verifying sequences of shRNA template.
Keywords |
| RNAi, cloning, reverse PCR, loop, vector, siRNA, shRNA, sequencing |
Introduction |
| RNA interference elicits posttranscriptional gene silencing in a sequence-specific fashion, which provides a powerful tool to explore a gene’s function. However, directly transfecting artificially synthetic siRNA duplexes results in temporal RNAi effects lasting only a few days in mammalian cells. To obtain long-term RNAi effects, the strategy to construct plasmid expressing short hairpin RNA has been developed (Brummelkamp et al, 2002), through which the shRNA-encoding cassette can be integrated into the host genome to produce shRNA continually. The intracellular apparatuses process the shRNA to form active siRNA. |
| To date, three main strategies have been described for establishing shRNA expression plasmids. The most common approach is to anneal two oligonucleotide segments containing the hairpin sequences and ligate this between two custom restriction sites at a vector (McManus et al, 2002). This strategy is employed for most commercial shRNA expression vectors. The second approach is to produce shRNA-encoding cassette by twostep PCR (Castanotto et al, 2002; Guo et al, 2003), and the PCR fragments are cloned into a vector. Primer extension is a third alternative method, by which two oligonucleotide segments overlapping at 3’end are annealed and the complete double-strand constructs are recovered by the 5’A3’ polymerase activity of DNA polymerase (Paddison et al, 2004; McIntyre and Fanning, 2006). A common drawback of all of the three strategies is the potential for forming a premature hairpin configuration, which we consider the dominant contributor to the failure of cloning. Hence, our proposal is to develop an alternative strategy bypassing premature hairpin configuration. |
| In this article we also discuss the issue that shRNAencoding cassettes containing mismatch nucleotides within antisense strand might invalidate its RNAi effects (Xia et al, 2003; Denti et al, 2004). The mismatch nucleotides might be introduced during plasmid construction, and empahsises the importance of verifying the sequence of shRNA-encoding cassettes. Unfortunately, sequencing reactions frequently fail to read out the whole shRNA cassette (due perhaps to selfcomplementary hairpin structure), which prompted us to consider the potential for inclusion restriction site in the loop so that DNA sequencing could be performed with regular linearized samples. |
Materials And Methods |
| Universe template for reverse PCR |
| A universal template used here refers to a plasmid containing a particular promoter for RNA transcription and related transcriptional terminator. To exemplify our strategy, we utilized the plasmid pGEM-H1 in this work. The plasmid pGEM-H1 was constructed by cloning a polIII H1 cassette into the pGEM-T vector (Promega). This H1 cassette was obtained from the plasmid psiRNA-hH1zeo (Invivogen) by PCR with the following primer pair: |
| Forward: 5’-GACCACGCGTATCGATACTA-3’ |
| Reverse: 5’- CTTTTGAAAAACCTCAGGTG-3’ |
| The following amplifications conditions were used: 94?for 20sec, 56?for 20sec and 72?for 20sec, for 25 cycles. |
| Cloning of short hairpin RNA cassettes |
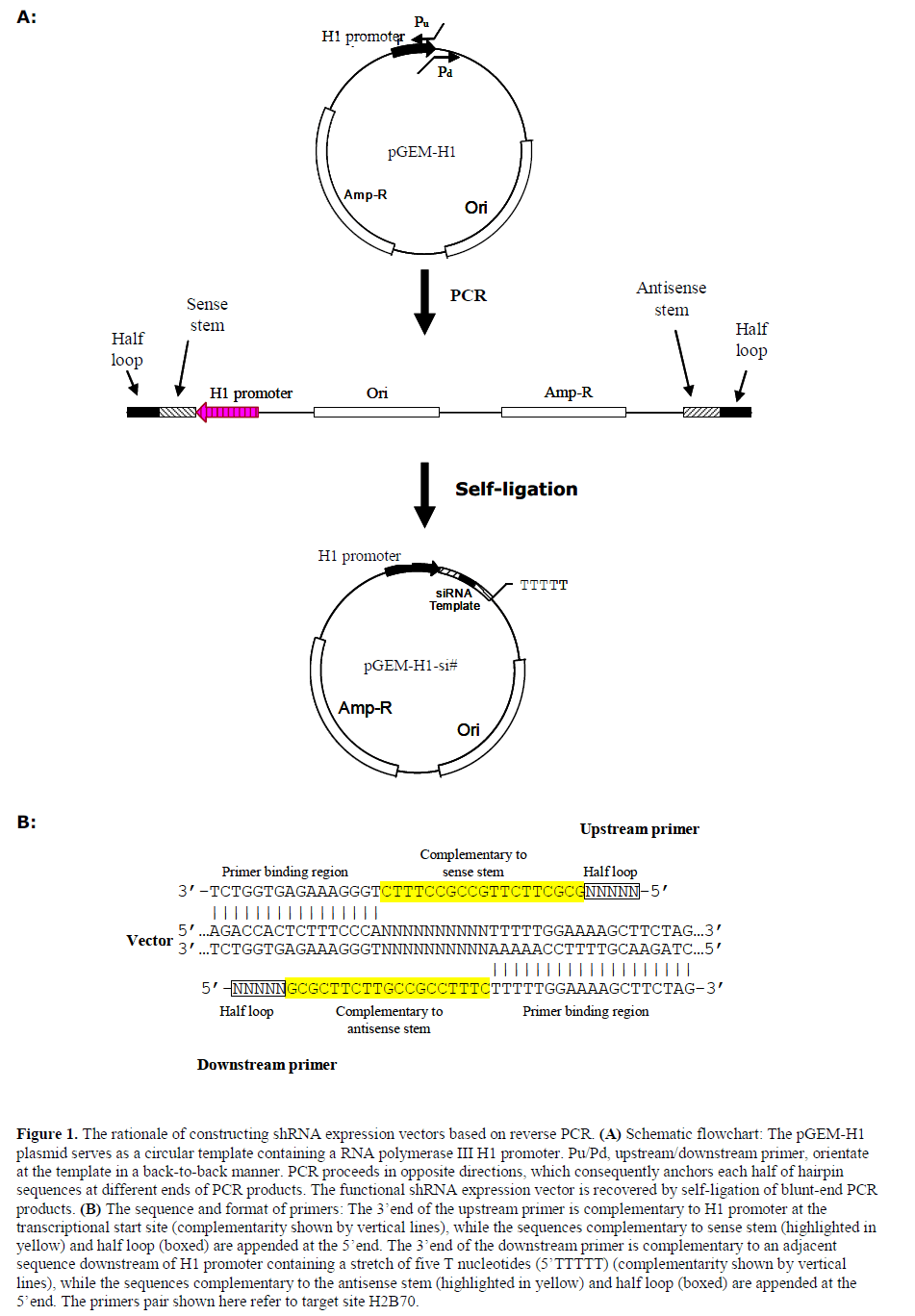
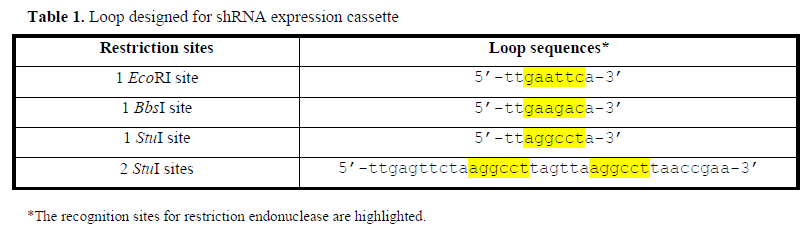
| We demonstrate the procedure of cloning shRNA cassettes targeting H2BGFP, a nuclear-localized fluorescent gene, at the site 5’-AAGAAAGGCGGCAAGAAGCGC-3’ that is located 70-nt downstream of the translational start codon of H2BGFP mRNA. This siRNA targeting H2BGFP gene had been validated previously (Huang et al, 2005; Huang et al, 2006). The cloning steps are schematically shown in Figure 1A. The 3’end of PCR primers complementary to pGEM-H1 plasmid, while the hairpin stem followed by half loop is appended at 5’ends (Figure 1B). Primer pairs used in this work varied the sequences corresponding to loop region in order to include restriction site (Table 1). The PCR reactions were carried out as follows: 94? for 30sec, 61? for 30sec, 72? for 90sec, for 28 cycles. The reaction was called ‘reverse PCR’ due to the primer pair binding to the template in a back-to-back orientation. |
| We used KOD plus DNA polymerase in his study (TOYOBO, Japan), which amplifies DNA with higher reliability than conventional Taq DNA polymerase and produces blunt DNA fragment without additional 3’-A overhangs. The purified PCR products were self-ligated by T4 DNA ligase (Promega). The trasnsformation of ligated DNA into E. coli host was carried out by routine methods (Sambrook and Russell, 1989). The positive clones were preliminarily identified with a single primer 5’- GCGCTTCTTGCCGCCTTTC-3’, which is cognate to antisense stem and hence capable of binding to both strands of a positive clone. |
Sequencing of siRNA template linearized by loop digestion |
| For each transformation, 5 preliminarily identified clones were amplified and plasmid extraction was performed using Wizard Plus Minipreps DNA Purification System (Promega). The plasmids were digested for 6 hr with appropriate restriction enzymes recognizing the loop sites present in these hairpins. DNA samples were directly analyzed in a 1% (w/v) agarose gel containing 0.5 μg/ml ethidium bromide by standard DNA electrophoresis. The confirmed linearized plasmid DNA was then isolated from gel and purified for sequencing using Wizard DNA Clean-Up system (Promega). DNA sequencing was performed with M13F and M13R primers by using ABI 377 DNA sequencer. |
Cell culture, transfection, and RNAi assay |
| The fluorescent Hela cell line which stably expresses H2BGFP reporter gene was established as previously described (Huang et al, 2006). Either 1μg of siRNA template plasmid or 1μg of a control shRNA-expressing plasmid (a scramble control shRNA expressing plasmid targeting to an irrelevant sequence 5'-AGATCTCAAG TTCCTCACA-3') was transfected into the fluorescent Hela cells with DOTAP (Roche) as described by the manufacturer. Cells were cultured in DMEM (Gibco) supplemented with 10% (v/v) fetal calf serum (Gibco), and harvested for analysis 48 hr after transfection. |
| FACS analysis was performed for the cells to quantitatively evaluating the down-regulation of fluorescence using FACSCalibur flow cytometer (BD Biosciences), and western blot analysis was performed using antibodies against GFP (Boehringer–Mannheim) as described previously (Huang et al, 2006). |
Result And Discussion |
| Cloning of shRNA based on reverse PCR products |
| shRNA expression plasmids are attractive as they may confer either long-term or inducible RNA interference. The elements corresponding to siRNA sense/antisense strand and loop are appended to the 5’end of PCR primers, respectively, while the 3’end is complementary to the universal template (Figure 1). The primer pair binds to the circular template back-to-back. As a result, the ready-to-ligate DNA segments that contained all necessary elements to form a functional plasmid are obtained through one round of PCR amplification. The establishment of circular plasmid is simply achieved by self-ligating the PCR products with T4 DNA ligase. Based on this strategy, we established a plasmid pGEMH1- H2B70si targeting H2BGFP gene. |
| Our approach has two primary advantages over previous reported methods; (i) only one step PCR reaction is needed prior to ligation; (ii) due to the distal orientation of primer pair, the sequences corresponding to sense strand and antisense strand consequently anchor at the two separate ends of PCR fragments, which will reduce the chance of premature hairpin configurations forming within the PCR products. The latter is judged to be especially beneficial to ensure effective PCR |
 |
 |
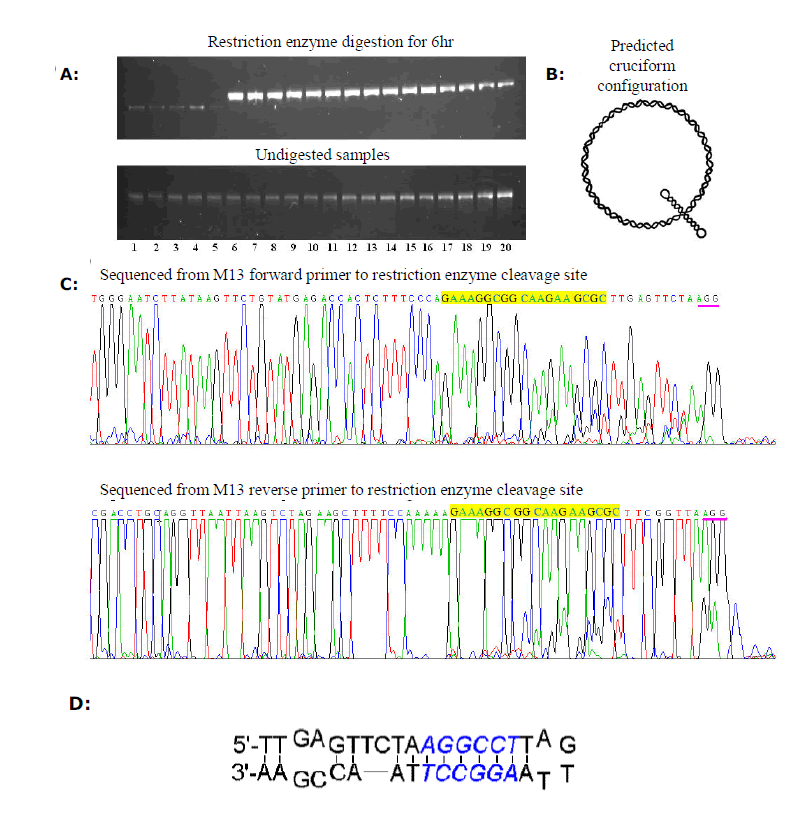 |
| Figure 2. Inclusion of twin restriction sites in the loop. (A) The loop including twin restriction sites of an shRNA expression plasmid is digested, while the loop including a single restriction site is resistant to digestion. Four groups of plasmids (each group consisting of five clones) that contained different loop sequences were digested for 6 hr with appropriate restriction enzymes. The electrophoretic analysis was performed in 1% (w/v) agarose gel for 30 min. Lanes 1-5; clones whose loop included twin Stu I sites: Lanes 6-10; clones with a single Stu I site: Lanes 11-15; clones with a single Bbs I site included; Lanes 16-20; cones with a single Eco RI site. (B) The predicted cruciform structure in a circular plasmid containing self-complementary regions. (C) The sequence of hairpin templates whose loop contains twin restriction sites can be determined on linearized segment in two separate reactions. The sequences corresponding to the hairpin stem are highlighted. The half-cleavage sites are underlined. (D) Prediction of the annealed configuration referring to the loop containing twin Stu I sites, which might form a digestible single-stand loop. Stu I recognition is shown as italics. |
| amplification in comparison with previous two-step PCR approaches, since the primer used in the second step will be inclined to anneal with the complementary fragment produced in the first step in term of two-step PCR approaches. The issue of premature hairpin configuration also affects strategies based on annealed oligonucleotide inserts, because intra-strand annealed by-products will compete with expected double-stranded products. |
| Sequence verification of siRNA templates |
| The structure of a shRNA-encoding cassette is characterized by a pair of inverted repeats, which enable the small RNA transcripts to fold back a short hairpin automatically. Unfortunately, hairpin template is a serious obstacle for DNA sequencing, which frequently results in abortion of sequencing reactions. Introducing a few mismatched nucleotides within the sense strand is reported helpful to alleviate sequencing difficulties (Yu et al, 2003; Miyagishi et al, 2004), but there has also been suggested a correlation between increasing mismatches number and decreased gene suppression activity (McIntyre and Fanning, 2006). As an alternative, we considered the strategy involving linearizing the vector by including restriction site(s) within the loop sequence worthy of investigation. |
| We tested four different loop sequences containing a unique Eco RI, Bbs I, Stu I, and twin Stu I, respectively. Stu I is used particularly due to the property that only one flank base is needed for recognition and digestion by restriction enzyme. The results indicated that only loop sequences including twin Stu I was cleaved, while all other loop sequences including a single restriction site were resistant to digestion (Figure 2A). Although these clones were not confirmed by sequencing the above results were sufficient to suggest that the loop including a single restriction site is resistant to digestion. The resistance we observed might be due to the configuration of plasmid in solution. The configuration of plasmid in solution fluctuates between regular circle and super-coil, and the stress present within super-coiled DNA molecules might lead to localized denaturation, especially for DNA segments containing self-complementary regions, and a cruciform loop configuration could be predicted (Watson et al, 1987). We speculate a shRNA expression vector might adopt a configuration as shown in Figure 2B and the loop region would be a single-strand bubble, which could account for the resistance of a single restriction site included loop because restriction enzymes recognize and digest double-strand DNA only. While the loop region including twin restriction sites is capable of forming intra-annealed “double-stranded” region, it is cleavable by a restriction enzyme. The plasmid pGEMH1- H2B70si, with a hairpin loop containing twin Stu I sites, could therefore be cut off at the loop region. |
| The linearized plasmid was sequenced in two separate reactions. The results are presented in Figure 2C, and sequences corresponding to sense strand and antisense strand fully sequenced, which demonstrated that inclusion of twin restriction sites in the loop is a practical strategy for verifying the complete sequences of a shRNA template. |
Validation of shRNA |
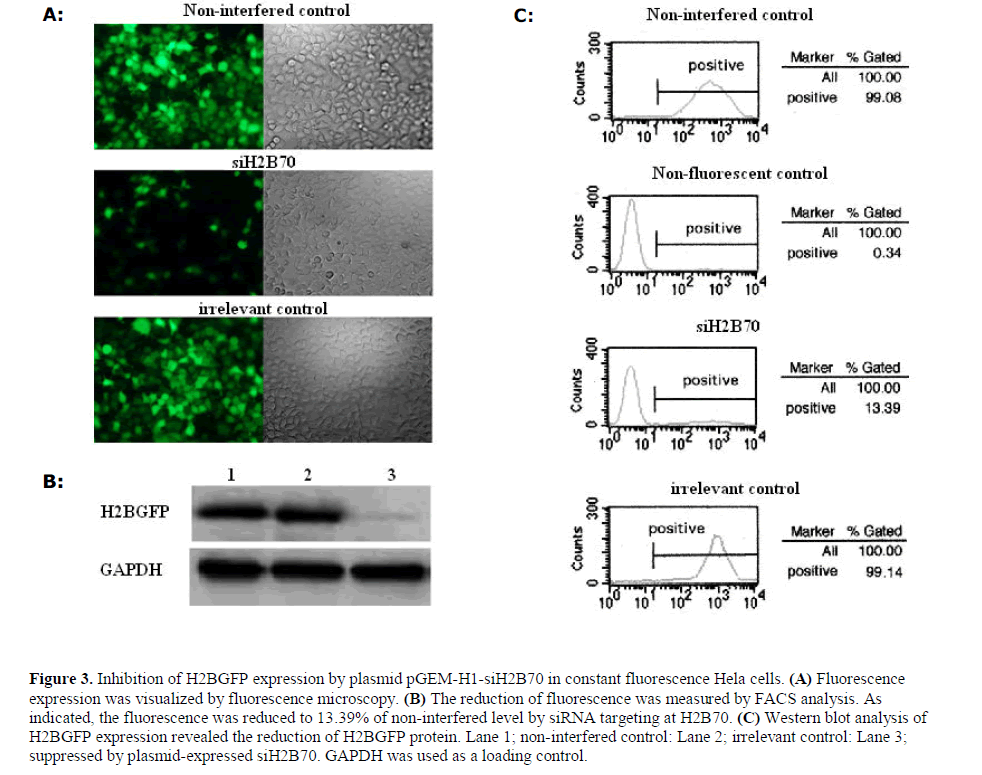
| The performance of plasmid pGEM-H1-H2B70si constructed using reverse-PCR was evaluated to prove the effectiveness of shRNA cassettes generated by this method. Fluorescent Hela cells that stably expressed H2BGFP were transfected with pGEM-H1-H2B70si, and the expression of H2BGFP quantified. The cell plates were first imaged in a fluorescence microscope, and then the cells were FACS sorted. The cell fluorescence images revealed apparent suppression of H2BGFP expression (Figure 3A), and the suppression of H2BGFP expression measured by FCAS analysis indicated reduction to 13% compared to an irrelevant control (Figure 3B). The expression of H2BGFP protein was probed anti-GFP antibody on western blot, and a suppression of H2BGFP at protein level was observed (Figure 3C). |
| Although, the loop of pGEM-H1-H2B70si containing twin Stu I sites is relatively long, the above observations demonstrated that such an shRNA construct is effective in eliciting gene suppression. In fact, this design is a mimic of Rnt1p-styled loop described by Bozzoni’s group (Denti et al, 2004) with a few altered bases to include twin Stu I sites. It is noteworthy that second generation small RNA hairpin vectors mimicking miRNA loop structures are more active that conventional shRNAs, suggesting that regions within the loop, or at the might influence DICER processing (Silva et al, 2005). |
Conclusions |
| The reverse PCR approach described in this paper provides a simple and fast strategy for constructing an shRNA expression vector, offering a one-step method over the previous PCR strategies that required two-step PCR amplification. In addition, we demonstrated that the loop of shRNA expression cassette including a single restriction site is resistant to restriction enzyme digestion, which is detrimental to sequence verification of such constructs. However, for those cases in which recovery of shRNA vectors from cells is crucially dependent on the ability to sequence the shRNA construct, the inclusion of twin restriction sites in the loop is a practical strategy for verifying the sequences of shRNA template. |
Acknowledgement |
| This study was supported by Shanghai Municipal Commission for Science and Technology (Grant No. 04DZ14006) and in part by E-institutes of Shanghai Municipal Education Commission (No.E03003). |
Statement Of Competing Interests |
| The authors declared no competing interests. |
 |
References
|