- Journal of RNA and Genomics (2007) Research Article
A novel expression system for artificial miRNAs containing no endogenous miRNA precursor sequences
| Atsushi Shibata*, Akiko Iwaki and Yasuyuki Fukumaki Division of Disease Genes, Research Center for Genetic Information, Medical Institute of Bioregulation, Kyushu University, Maidashi 3-1-1, Higashi-ku, Fukuoka 812-8582, Japan |
| Corresponding Author: Atsushi Shibata, Email: ash@gen.kyushu-u.ac.jp, Tel: +81 92 642 6168, Fax: +81 92 632 2375 |
| Received 19 February 2007; Revised 13 April 2007; Accepted 17 April 2007; Published online 27 May 2007 |
Abstract
Recently, artificial microRNA (miRNA)-mediated RNA interference (RNAi) systems have been developed as useful tools to study gene functions. We report an artificial miRNA precursor motif (AMPM) containing several restriction sites in the loop and in the extended stem structures, and generated a vector-based expression system for AMPM under the control of an RNA polymerase II promoter. The AMPM located in the intron or the exon of the selection marker gene mediated silencing of the reporter luciferase gene expression in HeLa cells. Northern blotting and colony formation assays revealed that the AMPM was efficiently and appropriately processed into mature miRNA. The AMPA system also down-regulated endogenous p53 and laminA/C proteins in stable transfectants. Moreover, clustered AMPMs separated by an appropriate spacer, targeting different sites within a single mRNA mediated gene silencing, even if the sequences of the target and the AMPM were partially complementary. This indicates the potential therapeutic utility of clustered AMPMs for highly-mutable targets, such as human immunodeficiency virus type 1.
Keywords |
| microRNA, miRNA, cluster, RNA polymerase II, HIV-1, gene therapy |
Introduction |
| RNA interference (RNAi) is now a powerful tool to suppress gene expression in various eukaryotic systems (Murchison and Hannon, 2004; Filipowicz, 2005; Sandy et al, 2005). In mammalian cells, chemically synthesized small interfering RNAs (siRNAs) and vector-based short hairpin RNAs (shRNAs), which are processed by a cytoplasmic endonuclease, Dicer, into siRNAs, have been used to mediate RNAi. Cells express small single-stranded RNAs 20-24 nucleotides (nt) in length, called microRNAs (miRNAs) (Lagos-Quintana et al, 2001; Lau et al, 2001; Lee and Ambros, 2001), which mediate gene silencing similarly to shRNAs (Bartel, 2004). miRNAs are encoded within intergenic regions and intronic regions of the eukaryotic genome, and are sometimes clustered (Altuvia et al, 2005; He et al, 2005). Initially, miRNAs are transcribed as a part of long precursor RNA (primarymiRNA or pri-miRNA) (Lee et al, 2002; Suh et al, 2004; He et al, 2005), and form a hairpin-like structure. A nuclear endonuclease, Drosha, processes this stem-loop structure into miRNA precursors (pre-miRNAs) ~70 nt long (Zeng et al, 2005b). The nuclear pre-miRNAs are exported to the cytoplasm by a nuclear transport receptor, Exportin-5 (Yi et al, 2003; Bohnsack et al, 2004), and processed by Dicer into mature miRNAs ~22 nt long. After incorporation into miRNA-containing RNA-induced silencing complexes, miRNAs bind to their target mRNAs with partial complementarity, and direct degradation of mRNA and/or translational suppression (Yekta et al, 2004; Bagga et al, 2005; Petersen et al, 2006). |
| Effective gene silencing in cultured cells (Zeng et al, 2002; Boden et al, 2004; Stegmeier et al, 2005; Zeng et al, 2005a; Zhou et al, 2005; Chung et al, 2006) and mice (Li et al, 2006: Rao et al, 2006; Xia et al, 2006) using expression vectors for artificially designed miRNA precursors is well documented. In these reports, expression vectors for synthetic miRNA precursors were constructed by substitution of the stem regions of natural miRNA precursors, such as miR-30 or miR-155, to fully match arbitrary target mRNAs, but the loop sequences remained intact. Here we designed a novel artificial miRNA, named AMPM, which incorporates restriction enzyme recognition sequences in the loop and stem region of the miRNA precursor structure. Despite the absence of any sequence of natural miRNA precursors, AMPMs were efficiently processed into mature miRNAs and suppressed the target gene expression. We also assessed the ability of dicistronic AMPM clusters targeting multiple sites within a single mRNA to suppress highly-mutable targets. The usefulness of AMPM as a gene silencing tool is discussed. |
Materials And Methods |
| Target site selection and AMPM design |
| The sequences of the GL3 firefly luciferase gene, the human p53 mRNA, the human lamin A/C mRNA and the HIV-1 genome were obtained from GenBank (GenBank accession Nos. U47296, NM000546, NM170707 and AF033819, respectively). The target site of the GL3 firefly luciferase included the target site of shRNA used in a previous report (Elbashir et al, 2001). The target sites of other genes were selected using the siRNA design tool freely provided by QIAGEN on the following website:- (http://www1.qiagen.com/Products/GeneSilencing/Custom SiRna/SiRnaDesigner.aspx). This design tool makes choices of 21 nt long target sites of siRNAs based on an original score. Endogenous miRNA precursors are processed at positions 22~26 nt from their stem-loop junctions by Drosha (Weber, 2005). We therefore selected 26-nt target sequences. The antisense regions of all AMPMs used in this study were designed to be fully complementary to their target mRNAs. The sense regions were modified to form a mismatch and a bulge structure in their stem regions and G-U wobble pairs in the 3’ half of the stem regions. Secondary structure of AMPMs was predicted using the program mfold (ver 3.2) (Zuker, 2003). (http://www.bioinfo.rpi.edu/applications/mfold/rna/form1.cgi). |
| Plasmid construction |
| Oligonucleotides used in plasmid construction are given in Table 1. To construct a parent vector for the AMPM cassette, the multiple cloning site (MCS) of pBluescript SK (+) (Stratagene) was replaced by a novel MCS in two steps. A pair of chemically synthesized oligonucleotides was first annealed to form MCS1 and inserted into the Kpn I-Eco RI site, and then MCS2 was similarly produced and inserted into the Eco RI-Xba I site of pBluescript SK (+). The resulting plasmid was designated as pBS-miR. To generate the AMPM-FL cassette, oligonucleotides corresponding to the antisense strand region (AMPM-FLA) and the modified sense strand region (AMPM-FL-mS) were inserted into the pBS-miR vector. After digestion with Pvu II and Eco RV, the AMPM-FL cassette was inserted into the blunted Bam HI site and the Nae I site in pVITRO1 (Invivogen), generating the exonic and intronic AMPM vectors, pAMPM-FL-exon and pAMPM-FLintron, respectively. Other AMPM vectors targeting the human p53 mRNA, the human lamin A/C, the HIV-1 integrase and the HIV-1 capsid genes were constructed in a similar manner using appropriate oligonucleotides given in Table 1. Human H1 promoter-driven shRNA vectors targeting the HIV-1 integrase and capsid genes, pH1-sh- IN1 and pH1-sh-CA1, were constructed using oligonucleotide pairs sh-IN1, and sh-CA1, respectively, as previously described (Brummelkamp et al, 2002) except that the stem regions we used were 26 bp long. |
| The clusters of AMPM cassettes were constructed in pBSmiR. A pair of oligonucleotides, INT, consisting of partial sequence of an interval sequence in the human miR-17 cluster on chromosome 13 (GenBank accession No. AB176708), and an Eco RV recognition sequence, was inserted into the Eco RV-Xba I site of pBS-AMPM-IN1. The Eco RV site was used for further concatenation of the interval sequence unit, which was 24 bp in length. The AMPM-IN1 cassette, obtained by digestion of pBSAMPM- CA1 with Pvu II and Xba I, was inserted into the Eco RV-Xba I site of pBS-AMPM vectors containing the AMPM-IN cassette and various lengths of interval sequences. The AMPM cluster vectors containing the AMPM-IN1 and AMPM-CA1 cassettes were constructed in a similar manner. |
| Series of reporter vectors, pFL-INs and pFL-CAs, were constructed as follows. Partial sequences encoding integrase (IN) and p24 capsid protein (CA) from several strains of HIV-1 were inserted into the Xba I site located in the 3’-UTR 7 bp downstream from the GL3 firefly luciferase open reading frame in the pGL3-Control vector (Promega). The IN1 and CA1 sequences were selected from an HIV-1 strain (GenBank accession No. AF033819). The sequences corresponding to IN1 from different strains of HIV-1, IN2, IN3 and IN4 were from GenBank accession Nos. AF484511, AY713418 and AF055729, respectively. The sequences CA2, CA3 and CA4 corresponding to CA1 were from GenBank accession Nos. AY134925, AY396897 and AY489925, respectively. To generate pFL-IN1-CA1, IN2-CA2, IN3-CA3 and IN4- CA4, oligonucleotides IN1, IN2, IN3 and IN4 were inserted into the Xba I site in the pFL-CA1, CA2, CA3 and CA4 vectors, respectively. |
| Cell culture and transfection |
| HeLa cells were grown in DMEM (Sigma-Aldrich) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (JRH Biosciences) and plated at 2 x 105 cells per well in 12-well plates or 4 x 105 cells per well in 6-well plates 24 hr prior to transfection. The cells were transfected using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer’s instructions. |
| To isolate stable transfectants, HeLa cells transfected with pAMPM-p53 or pAMPM-lamin A/C were replated in 100- mm dishes 24 hr post-transfection, and cultured with 200 μg/ml of hygromycin (Wako) for 2 weeks. Thereafter, colonies were counted, isolated and propagated. |
Northern blot analysis |
| The chemically synthesized DNA oligonucleotide probes to detect the antisense strand region of AMPMs are as follows: |
| AMPM-FL: |
| 5’-ATCACTTACGCTGAGTACTTCGAAATGT-3’ |
| AMPM-p53: |
| 5’-CTGGAAGACTCCAGTGGTAATCTACT-3’ |
 |
| AMPM-lamin A/C: |
| 5’-ACTGGACTTCCAGAAGAACATCTACA-3’ |
| AMPM-IN1: |
| 5’-AACAGGGCAGGAAACAGCATATTTTC-3’ |
| AMPM-CA1: |
| 5’-CCAGCGGCTACACTAGAAGAAATGAT-3’ |
| A probe for the modified sense strand region of AMPMFL was 5’-ACATTTCAAAGTACCAGCGGAAGTG-3’. These probes were labeled with O-32P ATP (MP Biomedicals) using T4 polynucleotide kinase (TOYOBO). HeLa cells were grown in 6-well plates and transfected with 8 μg of each plasmid per well. After 48 hr of transfection, the cells were harvested and used for total RNA extraction using Isogen (Nippongene). Twenty microgram total RNA was separated by electrophoresis in a 15% (w/v) polyacrylamide/8M urea gel, and electrotransferred onto a Hybond-N+ membrane (Amersham Pharmacia Biotech). After UV crosslinking, the membrane was hybridized with the 32P-labeled DNA oligonucleotide probe in Church solution (1 mM EDTA, 0.5 M phosphate buffer and 7% (w/v) SDS) at 37°C overnight. Washing was carried out at 37°C twice in 2 x SSC and 0.1% (w/v) SDS for 15 min. The signals were detected using the FLA- 8000 (Fujifilm). |
| Luciferase assay |
| HeLa cells were transfected in 24-well culture plates with 2 μg of either AMPM expression vector or mock vector, 0.2 μg of the pGL3-Control (Promega), pFL-IN1-4, pFL CA1-4 or pFL-IN1-4-CA1-4 reporter vectors, and 0.1 μg of the pRL-TK (Promega) transfection control vector. The cells were harvested at 48 hr post-transfection. The luciferase activities were measured with the Dual Luciferase assay kit (Promega) using a Lumat LB9501 luminometer (Berthold). The luciferase activity was defined as the ratio of the reporter firefly luciferase activity to the internal control Renilla luciferase activity. |
RT-PCR |
| Total RNA was extracted as described above. Reverse transcription was carried out using a GeneAmp RNA PCR kit (Applied Biosystems) with random hexamer primer according to the manufacturer’s instructions. Polymerase chain reactions were carried out with the following primers: 5’-TCTTTTTCGCAACGGGTTT-3’ and 5’- GACCTCCGGTCACCTATTCA-3’ for the hygromycin resistance gene (Hygr) (cycling parameters: 94oC for 2 min, followed by 28 cycles of 94oC for 20sec, 55oC for 10 sec, 72oC for 30 sec, and a final extension step of 72oC for 5 min) and 5’-GTCAGTGGTGGACCTGACCT-3’ and 5’- AGGGGTCTACATGGCAACTG-3’ for glyceraldehyde- 3-phosphate dehydrogenase (GAPDH) (cycling parameters: 94oC for 2 min, followed by 28 cycles of 94oC for 20sec, 60oC for 10 sec, 72oC for 30 sec, and a final extension step of 72oC for 5 min. PCR products were separated in a 1.5% (w/v) agarose gel by electrophoresis, and photographed after staining with ethidium bromide. |
| Western blot analysis |
| HeLa cells stably expressing AMPMs against endogenous targets, the p53 or lamin A/C gene, were harvested and lysed in SDS sample buffer. Cell pellets were homogenized by brief sonication and protein content was determined by modified Lowry’s procedure (Markwell et al, 1978). Twenty micrograms of total proteins were separated on 12.5% (w/v) SDS–PAGE gels and electrophoretically blotted onto Immobilon-P membranes (Millipore). Proteins were detected using specific primary antibodies, peroxidase-labeled secondary antibodies and the ECL Plus Western Blotting Detection System (Amersham Biosciences), and photographed using LAS- 1000 (Fujifilm). The primary antibodies were mouse antip53 monoclonal antibody (DO-1) (Santa Cluz), rabbit antilamin A/C polyclonal antibody (H-110) (Santa Cluz) and rabbit anti-GAPDH polyclonal antibody (FL-335) (Santa Cluz), which were used at dilutions of 1:400, 1:800 and 1:200, respectively. The secondary antibodies were horseradish peroxidase-linked sheep anti-mouse Ig and donkey anti-rabbit Ig antibodies (Amersham Bioscience), which were used at dilutions of 1:5000 and 1:8000, respectively. We used the immuno-reactivity of GAPDH as an internal control. We quantified the signals in the resulting blots by densitometric analysis using Image Gauge (Fujifilm). |
Results |
| Design of the artificial miRNA precursors and cloning of the AMPM cassettes |
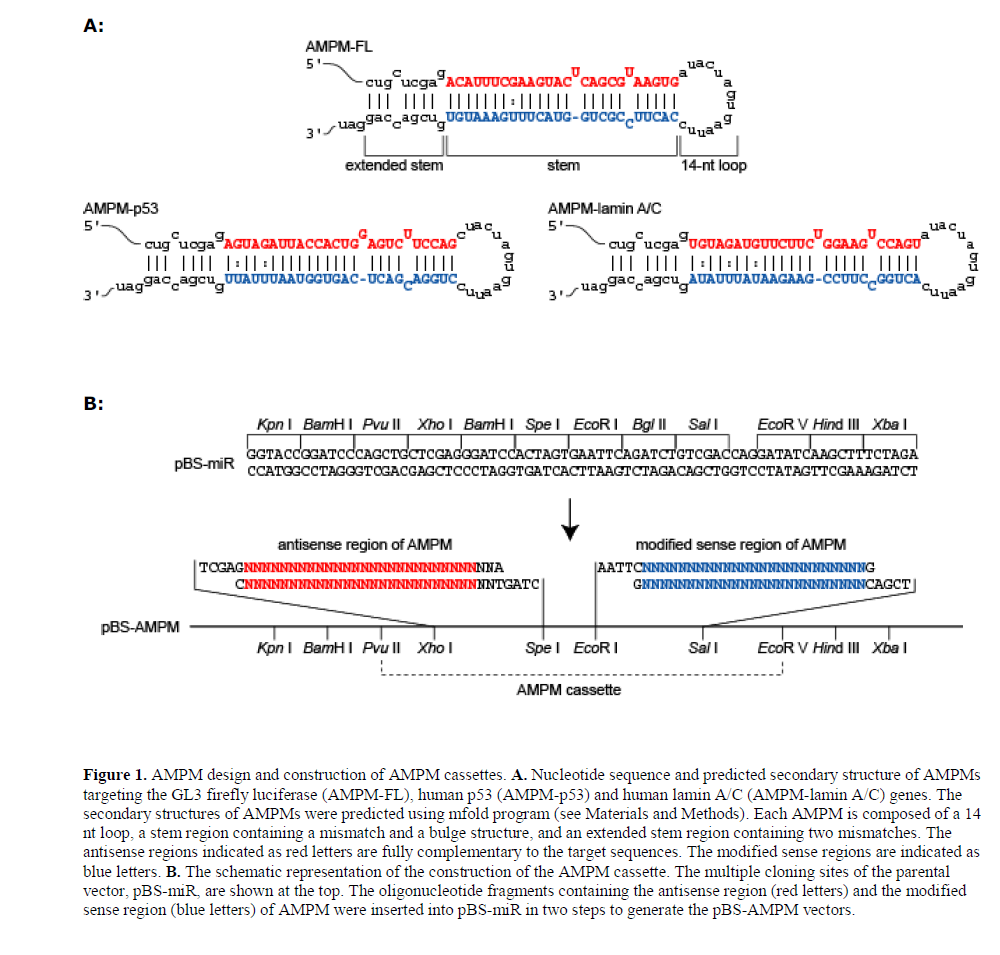
| AMPMs against the GL3 firefly luciferase (AMPM-FL), human p53 (AMPM-p53) and human lamin A/C (AMPMlamin A/C) genes were designed and their sequences and a predicted secondary structures are shown in Figure 1A. The target sites of AMPMs were selected as described in Materials and Methods. AMPMs were designed to mimic the secondary structure of the human miR-21 precursor (Zeng et al., 2005b) but they do not contain any sequence of miR-21 or any other endogenous miRNAs. Since a loop of >10 nt is essential for effective processing by Drosha (Zeng et al., 2005b), we used neighboring restriction sequences, Spe I and Eco RI, as the loop. The first two nucleotides of the loop structure were substituted to form a loop of 14 nt long in the mfold estimations. The antisense regions of AMPMs were designed to be fully complementary to the target sequences, but cytosine residues in the 3’ half of the sense strand regions were substituted by uridine residues to enhance asymmetric incorporation of the antisense strand into RISC, so called ‘strand bias’ (Khvorova et al, 2003). Additionally, a mismatch and a bulge structure in their stem regions were produced by modification of the sense strand sequences to mimic miR-21. At the terminus of AMPM, a part of the Pvu II and Xho I sequences at the 5’ terminus and the Sal I site and additional CAG sequences at the 3’ terminus form an extended double-stranded region. We confirmed the secondary structures of AMPM, including a mismatch and a bulge structure in the stem region and a loop of 14 nt by mfold analysis. |
| Prior to construction of the AMPM expression system, we prepared a cloning vector, pBS-miR, containing multiple cloning sites appropriate for the construction of an AMPM cassette. The oligonucleotides corresponding to the antisense region of the target gene and its modified sense region were introduced into the Xho I-Spe I and Eco RI-Sal I sites, respectively, generating pBS-AMPM (Figure 1B). |
| Construction of the artificial miRNA expression system |
| We generate an AMPM expression vector under the control of the EF1a promoter, we initially constructed an AMPM cassette targeting GL3 firefly luciferase (AMPMFL). The AMPM-FL cassette (Figure 1) was excised from pBS-AMPM-FL by digestion with Pvu II and Eco RV, and was then inserted into either the exon (5’-UTR) or the intron of Hygr in pVITRO1. The resulting plasmids were designated pAMPM-FL-exon and pAMPM-FL-intron, respectively (Figure 2A). |
| To assess whether AMPM was appropriately processed into the mature miRNA, pAMPM-FL-exon and pAMPM-FLintron were transfected into HeLa cells and the total RNA was isolated. Northern blot analysis showed a single band around 22 nt in length due to the expression of a shortly processed antisense region of AMPM-FL from both AMPM-FL vectors (Figure 2B). This data suggests that AMPM-FL is recognized as miRNA precursor and processed into mature miRNA, probably by Drosha and Dicer, regardless the location within a transcript. Moreover, a strand bias was observed because RNA molecule corresponding to the modified sense region could not be detected by northern blot analysis (data not shown). |
| When the AMPM-FL vectors were transfected together with the firefly luciferase reporter vector, luciferase assays showed that both vectors effectively suppressed the luciferase activity (Figure 2C), suggesting that both the exonic and the intronic AMPMs can mediate gene silencing. |
 |
| RT-PCR analysis showed that Hygr was expressed from the pAMPM-FL-intron as well as the empty control vector, but not from the pAMPM-FL-exon vector (Figure 2D). The expression of Hygr was further confirmed by colony formation assays. HeLa cells transfected with pAMPM-FL-intron normally formed colonies for 2 weeks under hygromycin selection, while no colonies were observed with pAMPM-FL-exon (data not shown). The ablation of the Hygr expression suggested the extremely effective processing of AMPM-FL located in the exon by Drosha in HeLa cells. Although northern blot analysis showed similar levels of AMPM-FL expression from the exonic and intronic AMPM-FL vectors, the processing of AMPM-FL in the intron did not affect the expression of Hygr, suggesting that the splicing event probably precedes the processing of AMPM-FL by Drosha. Therefore, we used the intronic AMPM system for further gene silencing experiments to achieve both effective gene silencing and expression of the selection marker gene. |
Silencing of endogenous genes by AMPM |
| We examined whether AMPMs could suppress the expression of endogenous genes. We used as targets the human p53 gene and the human lamin A/C gene, which were effectively suppressed in cultured cells by shRNAs (Brummelkamp et al, 2002; Sui et al, 2002). The intronic AMPM vectors targeting the p53 and the lamin A/C genes, pAMPM-p53 and pAMPM-lamin A/C, respectively, were each transfected separately into HeLa cells. After 2-week selection with hygromycin, independent clones of the cells stably expressing AMPM-p53 (Figure 3A) and AMPMlamin A/C (Figure 3C) were obtained. Western blot analysis showed significantly reduced expression of each target gene in these cell lines (Figures 3B and 3D), indicating that AMPM can silence the expression of endogenous genes. |
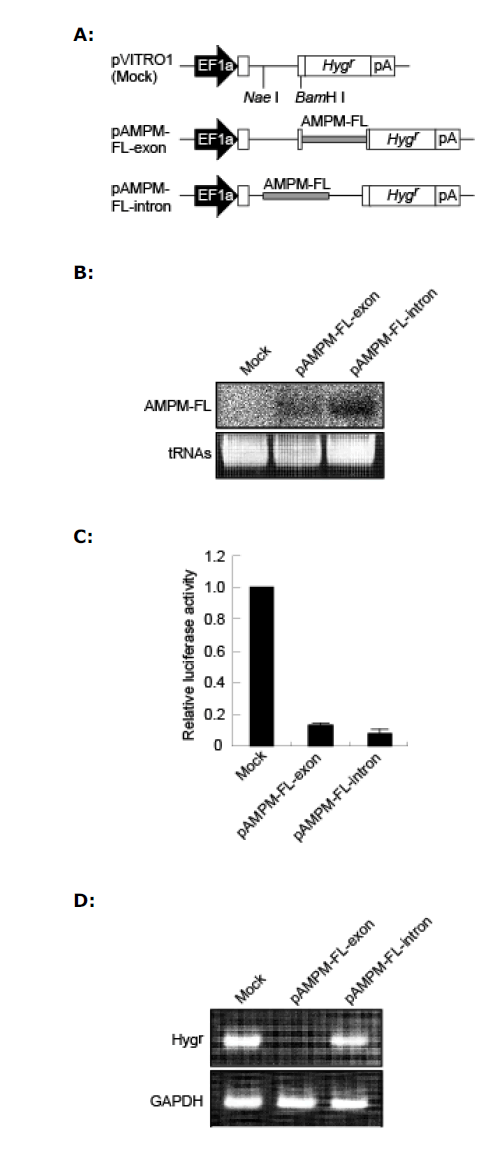 |
| Figure 2. Evaluation of AMPM expression systems by comparison of the level of expression of AMPM and of silencing efficacy. A. Schematic representation of the AMPM expression vectors. To generate the pAMPM-FL-exon and pAMPM-FLintron, the AMPM-FL cassette was inserted into the 5’-UTR and intron of Hygr, respectively, in pVITRO1. Gray boxes in the pAMPM-FL-exon and pAMPM-FL-intron indicate the AMPMFL cassettes. EF1a, the promoter of the rat translation elongation factor 1a gene; Hygr, the hygromycin resistance gene; pA, the human elongation factor 1a polyadeylation signal. B. The expression of a mature artificial miRNA targeting firefly luciferase. The pAMPM-FL-exon and pAMPM-FL-intron vectors were transiently transfected into HeLa cells and the total RNAs were isolated. Northern blot analysis using a radiolabeled oligonucleotide probe detected a band around 22 nt in length corresponding to the antisense strand region of AMPM-FL. Transfer RNAs (tRNAs) served as a loading control. C. Silencing efficacy of exonic and intronic AMPM-FL. The pAMPM-FLexon, pAMPM-FL-intron or pVITRO1 vector was transfected with the pGL3-Control reporter vector and pRL-TK normalization vector into HeLa cells. Normalized luciferase activities were standardized relative to levels in lysates from cells transfected with the empty vector, pVITRO1. The values are means with S.E.M. (n=3). D. The influence of the insertion of the AMPM cassette on the expression of Hygr. The pAMPM-FLexon, pAMPM-FL-intron or pVITRO1 vector was transfected into HeLa cells and the total RNAs were isolated 48 hours after transfection. RT-PCR was carried out with primers to detect Hygr and GAPDH mRNA. GAPDH served as a control. |
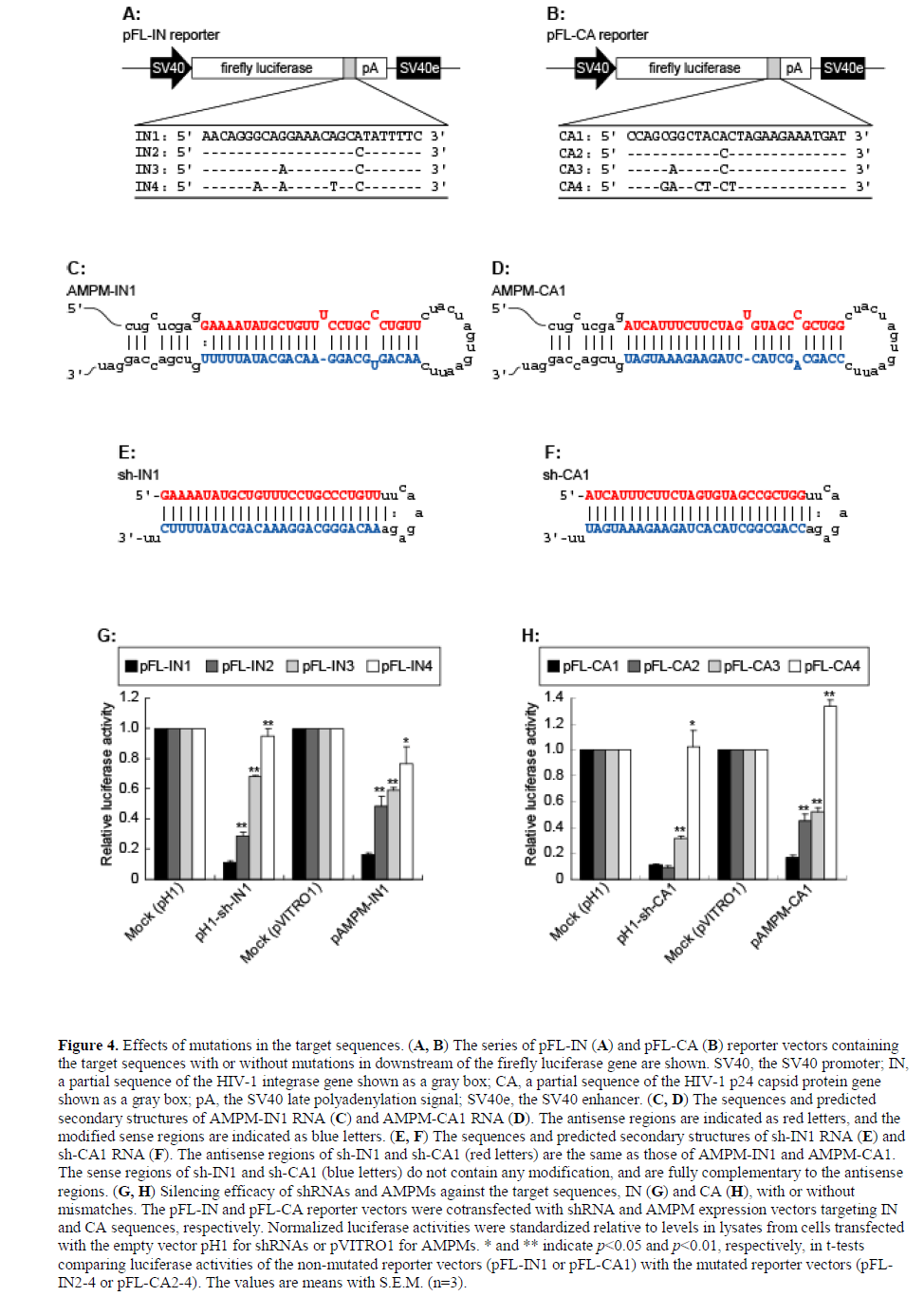
| Mutations in target sequences reduce RNAi efficacy of AMPMs |
| As natural miRNAs mediate RNAi despite their partial complementarity to their targets (Lagos-Quintana et al, 2001; Lau et al, 2001; Lee and Ambros, 2001), we assumed that AMPMs could mediate gene silencing even if the target sequences were mutated. HIV-1 is known to mutate easily, resulting in various mutant strains. When HIV-1 was targeted by RNAi in previous studies, HIV-1 could escape from RNAi by mutations in its RNA genome (Boden et al, 2003; Das et al, 2004). We therefore inserted partial sequences of the HIV-1 integrase (IN1) and capsid (CA1) genes and their mutant sequences derived from different HIV-1 strains (see Materials and Methods) into the 3’-UTR of the firefly luciferase gene (Figures 4A and 4B), and used them as reporter vectors to assess the sequence-specificity of AMPMs. The intronic AMPM vectors, pAMPM-IN1 and pAMPM–CA1, were constructed to express AMPMs fully complementary to IN1 and CA1 sequences, respectively (Figures 4C and 4D). Additionally, to compare AMPMs with shRNAs, human H1 promoter-driven shRNA vectors targeting the IN1 and CA1 sequences, pH1-sh-IN1 and pH1-sh-CA1, were constructed (Figures 4E and 4F). Luciferase assays showed that both AMPMs and shRNAs suppressed the reporter gene expression when the target sequences were fully complementary. Introduction of mutations in the target sequences attenuated the suppression of the luciferase activity according to the number of the mutations (Figures 4G and 4H), suggesting that AMPMs cannot silence the mismatched targets, like shRNAs. |
| Targeting multiple sites by the AMPM cluster overcomes effects of mutations in target sequences |
| How do natural miRNAs mediate RNAi effectively despite their partial complementarity? We next took note of the fact that the target sites of natural miRNAs were often redundant. We assumed that AMPMs could overcome the effects of mutations in RNAi by targeting multiple sites within a single mRNA. To examine this possibility, we constructed a series of reporter vectors that contained both HIV-1 IN1 and CA1 or their mismatched sequences in the 3’-UTR of the firefly luciferase gene (pFL-IN1-CA1, IN2- CA2, IN3-CA3 and IN4-CA4) (Figure 5A). The design of these constructs was based on the fact that the HIV-1 integrase and capsid proteins are encoded in a single transcription unit of the HIV-1 genome. |
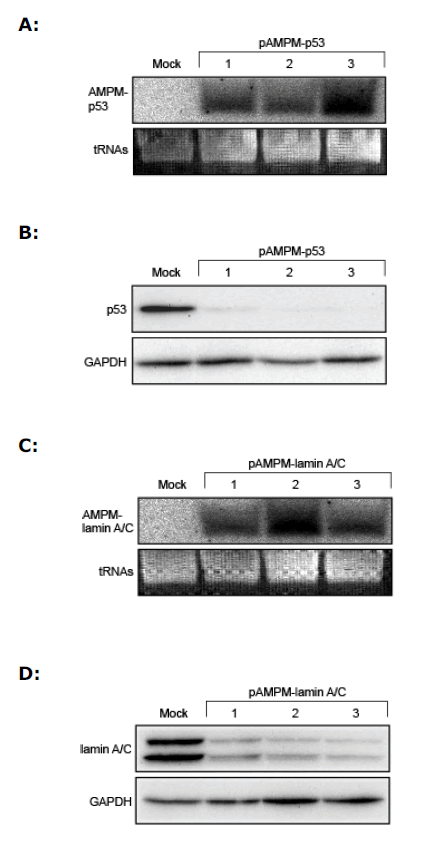 |
| Figure 3. Stable gene silencing of endogenous genes by AMPMs. (A, C) Stable expression of AMPMs targeting endogenous genes. The total RNAs were isolated from stable transfectants of pAMPM-p53 (A) and pAMPM-lamin A/C (C). Northern blot analysis showed the expression of mature artificial miRNAs targeting p53 and lamin A/C as a single band around 22 nt in length. tRNAs served as a loading control. (B, D) Western blot analysis for p53, lamin A/C and GAPDH in cells stably expressing AMPM-p53 (B) and AMPM-lamin A/C (D). Lanes 1 to 3 contained samples from independently isolated cell lines. GAPDH served as a loading control. |
| The AMPM-IN1 and AMPM-CA1 cassettes were clustered with interval sequences of various lengths (Figure 5B), and the resulting AMPM cluster vectors were transfected into HeLa cells and total RNAs were isolated. Northern blot analysis showed that mature artificial miRNAs for IN1 and CA1 were produced from pAMPM-IN1-48-CA1 and pAMPM-IN1-96-CA1 as well as the monocistronic pAMPM-IN1 and pAMPM-CA1vectors, while the products from pAMPM-IN1-0-CA1 could not be detected (Figure 5C). The level of IN1 miRNA expression from pAMPMIN1- 24-CA1 was relatively low, although that of CA1 was almost the same as pAMPM-CA1. These results indicate that the length of the interval sequence between two AMPMs can affect the processing of AMPMs. |
| AMPM cluster vectors were co-transfected with the various pFL-IN-CA reporter vectors into HeLa cells (Figure 5D). When we used pFL-IN1-CA1 as a reporter, the AMPM cluster vectors with interval sequences of 24, 48 and 96 bp showed enhanced gene silencing in comparison with the monocistronic pAMPM-IN1 and pAMPM-CA1vectors. It is noteworthy that the dicistronic pAMPM-IN1-48 and 96- CA1 vectors showed effective suppression even against mismatched target sequences, such as pFL-IN2-CA2 and pFL-IN3-CA3. These results indicate that targeting of multiple sites within a single mRNA by clusters of different AMPMs with an interval sequence of more than 48 nt could overcome the effects of mutations in target sequences. Therefore, this AMPM technology can be a powerful tool for gene therapy against viruses such as HIV-1, which often acquire drug resistance via mutations. |
Discussion |
| We have shown here that our artificially designed miRNA precursor, AMPM, that contains restriction sites in the loop and in the terminus of the stem region within the miRNA precursor motif, is useful as a gene silencing tool. Although AMPMs do not contain any sequence from natural miRNA precursors, they are effectively processed (perhaps by Drosha and Dicer), and the resultant short products can mediate effective gene silencing. Furthermore, the intronic AMPM system enables coexpression of artificial miRNAs and a selection marker gene. Although AMPM cassettes have to be cloned into pBS-miR prior to construction of AMPM expression vectors, this step makes the AMPM system flexible for application to various expression systems. Actually, we could easily insert AMPM cassettes into different vectors and observed coincidence of the expression of a fluorescent marker protein and effective gene silencing in several cell lines (data not shown). |
| The presence of mutations in the target sequence easily prevents suppression of the target gene expression by both monocistronic AMPMs and shRNAs. This observation indicates that AMPM technology can be applied to gene function analysis and gene therapy in a sequence-specific manner, like the shRNA system. We noticed that mutations in the CA target sequences affected silencing efficacy less than those in the IN target sequences. This may be due to the location of the mutations within the target sequences. In the CA target sequences, the core sequence for miRNA-mRNA interaction, located in the 3’ half of the target sequences, is conserved, suggesting that the “seed” sequence of AMPM-CA1 is preserved (Tomari and Zamore, 2005). |
 |
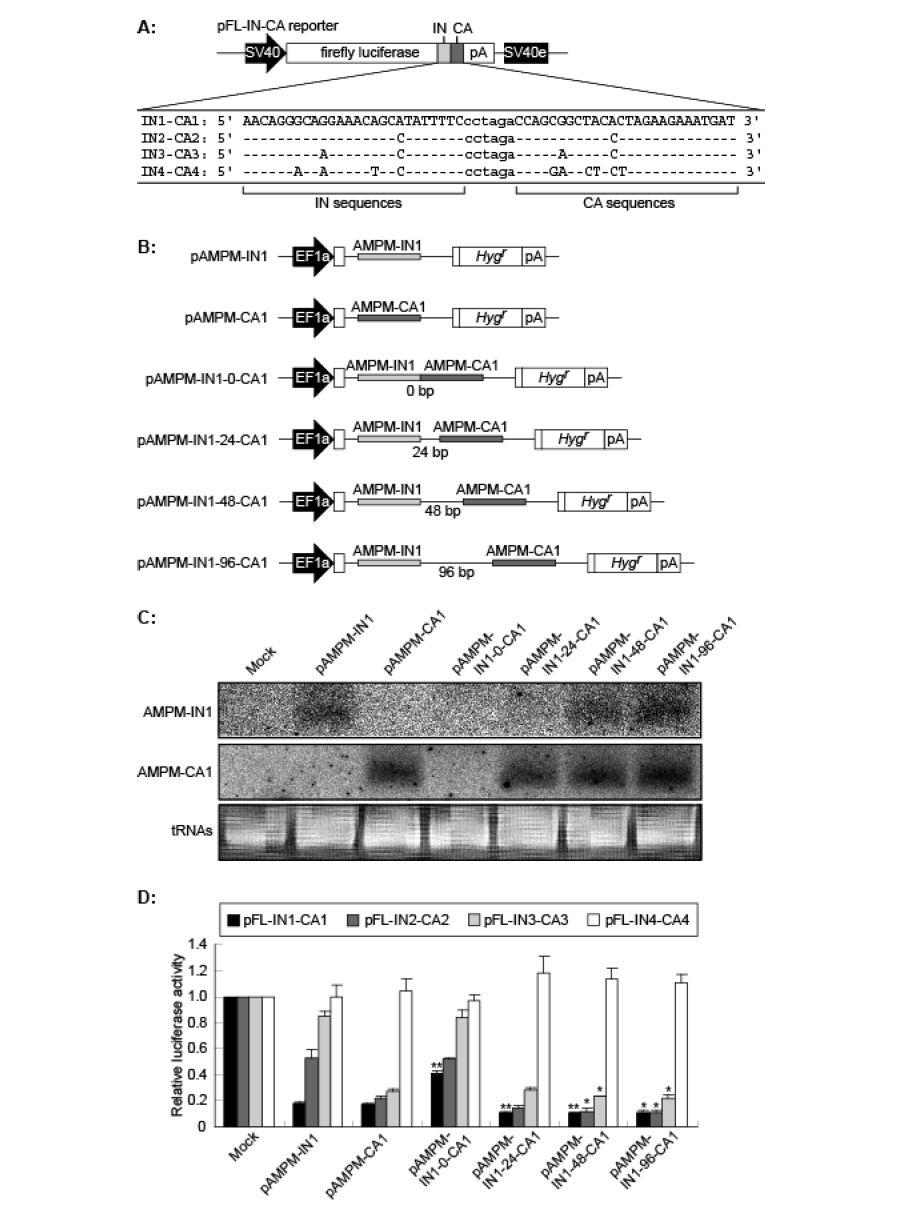 |
| Figure 5. The AMPM cluster vectors targeting different sequences within a single mRNA and their silencing efficacy. A. The tandemly arranged target sequences are shown as capital letters below a schematic representation of the reporter vector. The IN and CA target sites were linked via the interval of 6 nt long. SV40, the SV40 promoter; IN, a partial sequence of the HIV-1 integrase gene shown as a light gray box; CA, a partial sequence of the HIV-1 p24 capsid protein gene shown as a dark gray box; pA, the SV40 late polyadenylation signal; SV40e, the SV40 enhancer. B. Schematic representation of the AMPM cluster vectors with various lengths of interval sequences. The AMPM cassettes targeting the IN1 and CA1 sequences are shown as light gray and dark gray thin boxes, respectively. EF1a, the promoter of the rat translation elongation factor 1a gene; Hygr, the hygromycin resistance gene; pA, the human elongation factor 1a polyadenylation signal. C. The expression of mature artificial miRNAs from AMPM cluster vectors. The monocistronic AMPM vectors and AMPM cluster vectors were separately transfected into HeLa cells and the total RNAs were isolated. Northern blot analysis showed the shortly processed RNA derived from antisense strand region of AMPM-IN1 (top) and AMPM-CA1 (middle) around 22 nt in length. tRNAs served as a loading control. D. Silencing efficacy of each construct. The AMPM cluster vectors were co-transfected with the pFL-IN-CA reporter vectors. Normalized luciferase activities were standardized relative to levels in the lysate from cells transfected with the empty vector pVITRO1. * and ** indicate p<0.05 and p<0.01, respectively, in t-tests comparing the AMPM cluster vectors with the monocistronic AMPM vectors. The values are means with S.E.M. (n=3). |
| In the AMPM cluster system, the interval sequence between two AMPMs affect the efficacy of RNAi. An interval sequence of 24 nt between two AMPMs was sufficient to mediate RNAi, although interval sequences of 48 nt and 96 nt were more efficient. Northern blot analysis showed the maximum production of mature artificial miRNAs when the interval sequences were more than 48 nt in length. These observations are consistent with the fact that the interval sequences between the nearest miRNA precursor motifs in the human miR-17 cluster (GenBank accession no. AB176708) are around 30-60 nt long. This may reflect the efficacy of recognition and processing of AMPM by Drosha. Indeed, Zeng and Cullen reported that flanking non-structured sequences are essential for efficient processing of pri-miRNA by Drosha (Zeng and Cullen, 2005). |
| We also showed that use of the AMPM cluster system overcame the effects of mutations in the multiple target sites within a single mRNA (Figure 5), suggesting the possible therapeutic usefulness of the AMPM technology against highly mutable targets such as HIV-1. Natural miRNAs often bind to clusters of repetitive target sites with partial complementarity to each other (Lagos- Quintana et al., 2001; Lau et al., 2001; Lee and Ambros, 2001), suggesting that miRNAs accomplish the fine-tuning of gene expression by the binding of multiple sites with partial complementarity to target mRNAs. |
| The AMPM system can also be used to temporally and spatially suppress gene expression based on the fact that it is a Pol II-driven system. A recent study suggested that excessive shRNA in cells causes oversaturation of endogenous miRNA pathways and often results in lethality in mice (Grimm et al., 2006). The AMPM system may prevent such side effects because the transcriptional levels of AMPMs could be controlled by the use of appropriate Pol II promoters. |
Conclusions |
| • A newly designed artificial miRNA, named AMPM, can be efficiently processed perhaps by Drosha and Dicer, despite the absence of any sequence of natural miRNA precursors. |
| • AMPM can accomplish stable silencing of endogenous target genes in transfected cells. |
| • AMPM clusters targeting multiple sites within a single transcript can overcome mutations in the target sequences and be applied for the suppression of highly mutable targets such as HIV-1. |
Acknowledments |
| This work was supported in part by the 21st Century COE Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan. |
Statement Of Competing Interests |
| The authors declared no competing interests. |
List Of Abbreviations |
| AMPM: Artificial miRNA precursor motif |
| MCS: Multiple cloning site |
| Hygr: Hygromycin resistance gene |
| GAPDH: Glyceraldehyde-3-phosphate dehydrogenase |
| HIV-1: Human immunodeficiency virus type 1 |
References
|