Research Article - Biomedical Research (2017) Volume 28, Issue 8
A light weight SNP detection algorithm for the breast cancer targeted sequencing data
Guobin Chen and Xianzhong Xie*Institute of Personal Communications, Chongqing University of Posts and Telecommunications Chongqing, PR China
- *Corresponding Author:
- Xianzhong Xie
Institute of Personal Communications
Chongqing University of Posts and Telecommunications
PR China
Accepted date: December 30, 2016
Abstract
Many methods have been developed for Single Nucleotide Polymorphism (SNP) detection, but these methods are based on BWA or Bowtie matching software, because of the existence of this high false positive software, resulting in the test results Incorrect. The sequencing target region covered the entire coding region, exon-intron junction region (20-50 bp) and partial intron region of BRCA1/2 genes, 328 exon, 101.3k regions (http://pan.baidu.com/s/1kVNlWQb). A SNP detection algorithm based on breast cancer target DNA sequencing data is proposed, which has more advantages than Bcftools and IGV, which are the most commonly tools.
Keywords
SNP, Breast cancer, Sequencing
Introduction
Detecting genetic variation in the human genome is a crucial reason for understanding the phenotypic variation, including susceptibility to cancer and infectious diseases. Single Nucleotide Polymorphisms (SNPs) are one of the most common types of genetic variation in humans. SNPs influence protein compilation [1], transcriptional regulation [2], alternative splicing [3] and non-coding RNA regulation [4]. In general, SNPs of about 3.6 to 4.4 M SNPs are found in one human genome, and SNPs of high frequencies (>95%) are higher in SNPs in dbSNP. Are generally not the major mutation sites of disease? With the emergence of second-generation sequencing technology, sequencing costs and the emergence of high-performance computer, makes the whole gene sequence to find SNP possible.
At present, SNP methods for NGS data discovery are MAQ [5], SOAPsnp [6,7], SNVmix2 [8] and Bcftools [9], GATK [10], MAQ and SOAPsnp are short sequence alignment tools that uses the mass fraction deduced sequence and the alignment of alignments, and the MAQ makes full use of the pairing information to estimate the probability of each alignment read error, and also uses the Bayesian statistical model to evaluate the final Genotype error probability, and The mixed binomial model is used to discover the SNP for SNVmix2, giving the confidence score for each SNP that is invoked. While Bcftools and GATK use Samtools as the basis for estimating SNPs using Bayesian likelihood estimates. The above methods are derived by establishing a probability model, are BWA or Bowtie and other software to match the sequence, the sequencing sequence alignment to the human genome location hg19, and then on the Sam file related operations. First of all, BWA or Bowtie comparison is currently relatively good comparison of the software, but there are many problems [11], such as: false positive, false negative, reverse sequence cannot match the problem; 16G memory can run more problems. There are a number of SNP points that cannot be found by manipulating the Sam file after the software. It is possible to design a fast and accurate SNP algorithm for breast cancer targeted sequencing data, which is based on exon sequencing data of 20 genes, with small amount of data and high reproducibility between the data for breast cancer targeting DNA sequencing data. Based on the characteristics of breast cancer targeting DNA sequencing data, this paper proposes a SNP detection algorithm based on position index. The location accuracy of SNP point is higher than BWA based software.
Materials and Methods
Breast cancer targeted DNA sequencing data
The most common sequencing strategies for breast cancer, The sequencing of the genesare BRCA1/2 [12], RAD51C [13], BAD50 [14], MLH1 [15], BARD1 [16], ATM [17], STK11 [18], PTEN [19], TP53 [20], PALB2 [21], CDH1 [22], BRIP1 [23], CHEK2 [24], TLR4 [25], MAP3K1 [26], FGFR2 [27], TOX3 [28], LSP1 [29], and CCND1 [30] are used for DNA sequencing. The sequencing target region covers the entire coding region, exon-intron junction region (20-50 bp) and partial intron region of BRCA1/2 genes, with 328 exon and 101.3k regions.
The sequencing data of the target DNA sequence were Fastq type, double-ended sequencing, mainly concentrated in the exon region of the gene, the sequencing sequence was relatively short, there was overlapping phenomenon for the double-ended sequencing sequence data; for most of the sequences from a point. The sequencing sequence has a large number of repeat sequences; there is a large number of non- DNA sequences in the sequencing sequence, but not the common sequence and the linker sequences of the sequences are successfully captured; a large number of index sequences exist in the sequencing sequences and 5' and 3' end is not high quality bp.
Data cleaning
The original data of DNA target sequencing need to be cleaned up before proceeding to the next step. The structure of the library to be measured and the index and R of the sample file.
Index and R fine structure (Note: PE1.0 to PE2.0 method, from 5' to 3' end, black sequence index sequence, Index structure for the 5-8 base +19 base fixed fragment, sample Single-ended index, in the index were named index and R close to PE2.0, for example:
ACGTGTTACGTAATCGGGAAGCTGAAG
TATCCAGCCGTAATCGGGAAGCTGAAG
TGCAGTTCGTAATCGGGAAGCTGAAG
PE 1.0 and PE 2.0 are Illumina's PE PCR Primer 1.0 and PE PCR Primer 2.0 in the following sequence:
PE1.0:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCT ACACGACGCTCTTCCGATCT
PE2.0:
CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATT CCTGCTGAACCGCTCTTCCGATCT
PE1.0, PE2.0 for a fixed length, you can directly clean up the original data, clean up the original data only Target DNA and index data.
Index data cleaning
Index data sequence in another reverse-sequence file of bidirectional sequencing, the index sequence begins with all clean-up, where mismatch is also considered.
DNA sequencing, the sequencing company based on different samples sequencing index sequence, when the target sequence is ideal, the joint sequence will not affect the target sequence; if the target sequence is relatively short, another segment may be sequenced to index part of the sequence, so that Resulting in sequence S2 in the 3' segment sequencing data and index end sequence coincidence.
In this paper, SNP detection method, you must clear this part of the sequence, otherwise the Part index data will produce more SNP points, the reason is that this Part index caused, Described in R.
For (i in 1: length (sequence)) {pos=match Pattern (index, substr (sequence, 1, nchar (cha)+2), max. mismatch=3)}
if (length (width (pos))!=0) {sequence=substr (sequence, end (pos)+1, nchar (as.character (sequence))) }
The algorithm takes into account that the index has the wrong match problem and set to 3.
Common sequence data cleaning
A common sequence is used to capture a DNA sequence. If the DNA sequence is not successfully captured, a common sequence is considered to be a DNA sequence. If the capture is successful, no common sequence will appear in the original sequence. Common sequences capture DNA sequence shown in Figure 1.

Figure 1: DNA sequence capture classifications.
As can be seen from Figure 1A above, the DNA sequence has been successfully captured and the Target DNA sequence will not contain a Public sequence of 150 bp in length. In Figure 1B, the DNA sequence is captured by using a common sequence. If there is a common sequence in the sequence, it indicates that the sequence is not captured. In the double-ended sequencing, two common sequence cross-captures, a segment may contain a target sequence, and another segment may contain a common sequence, as shown in Figure 1C. Clean up the common sequence is to obtain better sequencing sequence, improve the efficiency of alignment. For the above-mentioned situation should be cleaned up, clean up the following principles:
1) If both ends of the sequence contain a common sequence, then both ends of the sequencing data should be cleaned up;
2) If one end of the sequence contains a common sequence, the other side contains the target sequence, then both ends of the sequencing data should be cleaned up.
Results
DNA sequence matching algorithm based on hash position index
In the DNA sequence, the key is how to achieve fast matching to the target sequence sequencing, taking into account the short target sequence, sequencing sequence repeatability is high, a hash index based DNA sequence matching algorithm, and the proposed method allows mismatching.
Hash table index
In order to improve the speed of DNA matching index, in order to be able to access any index entry in the effective time, the indexed DNA sequence is stored in the hash table. In order to map the sequence of DNA sequencing quickly to the location in the hash table, each character of the DNA base is mapped to an integer and the mapping values of any two characters are not repeated. The map function Map (x), The four bases in the DNA {A, C, G, T}, size 4, the Map (x) range from 1 to 4. The Map (x) is function mapping.
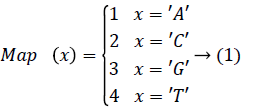
To determine the mapping function Map (x) for each character in the base table, we need to calculate the hash value for each DNA sequence, i.e. the corresponding storage location. If x='AA', then Map (x)=1; If x='TT', then Map (x)=16.
Target sequence position index
In the sequence range, it is the key method to find out the position of the sequence from the sequencing sequence, and allow the existence of mismatch (less than 3 mismatches in this paper). If it is more than 3, the quality of sequencing sequence is not very high, the entire sequence can be lost. If you compare all the sequences, you can set the mismatch to 1; all sequences can be retained, so the computer running time may be longer. If we consider the presence of InDel in the sequence, we can see that the high frequency sequence is not participating in the comparison after the sequence comparison. If there exists such sequence, it shows the existence of InDel.
The target sequence is set up according to the length of the actual target sequence. The objective sequence is short, and two bases are chosen as the index position to construct the table. If the target sequence is indexed, Target sequence is too long, for example: chr1 as the target sequence, then the establishment of four bases or eight bases as the index position to build the table. The method is as follows: Scan the target sequence to find out the location of all the AA, there is a list in the hash table position, the position relationship is a variable length array; sweep to TT all positions, there is a hash table 16 list position.
Sequencing sequence position
After indexing the position sequence of the target sequence, scan the sequencing sequence, and use the index relation of the hash table to map the sequence to the position relation. According to the position relation, there are P1 → P2 → ...→ Pk (k is the target sequence length half), allowing there are three mismatches between P1 → P2 → ..., → Pk. If present, the position is output, and if not, the next sequence is selected.
If the sequencing sequence has a positional relationship, the positional relationship Pos (i) is as shown in Equation 2,
Pos (1)=Pos (i)-(i-1) × 2 → (2)
For example, if ACTC is searched, AC={4, 9}, TC={3, 6}, AC and TC location index can be related. ACTC location is 4 in the target index.
SNP discovery algorithms for targeted DNA sequencing
Since sequencing of the exon of a specific gene by targeted sequencing has a shorter sequencing sequence and a deeper depth of sequencing (the test depth is 1000), the range of sequencing sequences is shown in the Table 1.
| NO | chr | Start | End | Width |
|---|---|---|---|---|
| 1 | chr2 | 215593342 | 215593770 | 429 |
| 2 | chr2 | 215595089 | 215595279 | 191 |
| 3 | chr2 | 215609736 | 215609926 | 191 |
| …. | ||||
| 327 | chr22 | 29126380 | 29126542 | 163 |
| 328 | chr22 | 29130300 | 29130749 | 450 |
Table 1: Target DNA sequencing target region.
In the table above, width contains the exon and part of the intron, width is the sequencer sequencing width, Illumina sequencing sequence length of 150, because it is double-ended sequencing, when the width of the exon is very short, Sequencing will produce overlap phenomenon. In this paper, SNP discovery algorithm is designed according to the characteristics of the targeted sequencing sequences. Most of the exons are only about 100 bp in length. When double-ended sequencing, S1 is generally sequenced from the start position and sequenced several times. 1000), S2 from the end position of sequencing, S1, S2 position the average number of sequencing of 1000, this will inevitably lead to a large number of repetitive sequences. SNP algorithm is based on a large number of repetitive sequences in the sequencing process, the frequency of repeated sequences for statistics.
Here are the specific algorithm steps:
1. Clean up the S2 sequence of the index sequence index, different sample index is different, all the sample index to clean up, due to 5' sequencing error, some index is the beginning of the beginning of N, the index of the first letter when N treatment;
2. Clean up some of the index in the S1 sequence, the target width less than 150 bp, first clean up the S2 end of the index sequence, and then through the S2 sequence information to find the S1 end, the end of the end of several bp and index to match, matching successful to clean up;
3. Clear the scope of the target is not captured data, S1, S2 common sequence search, if found to the public sequence, the data on both ends should be cleaned up;
4. The two-side data for statistical correlation frequency and sorted by frequency in descending order;
5. Select the relevant frequency of the data to compare, in theory, the sequences appear less frequent sequences are less, due sequencing data quality, there will be some errors in the sequence.
6. The use of hash indexing DNA sequence matching algorithm, select the range of the target range and meet the frequency of the data to compare, S2 in the data inversion, allowing mismatch is less than 3. If the data in S1 or S2 are not aligned with the target range, reverse the comparison again. The results of the comparison mainly record the position and frequency of the subsequence in the target sequence.
7. The range of the target sequence comparison of the sequence, starting from the location and range of the target range of one by one comparison, the statistical sequence number of bp and the target sequence cannot be compared to the number of times.
8. For the ratio of bp cannot be compared with the total number of times, the ratio is greater than 0.05, the output sequence SNP point sequence of chromosomes, position, target sequence in the original bp, bp after mutation, single bp mutation ratio.
Through the above steps can be achieved from the original sequence and the target sequence related operations, the direct output SNP sites.
Data analysis
Existing SNP methods are based on the BWA and Bowtie software such as the Sam file, use Samtools on the Sam file sort, sorted file Bam file, the use of Samtools in the mpileup command to generate bcf file. Then use bcftools for SNP analysis. Bcftools is included with Samtools software, generate a bcf file there are a large number of SNP points to be selective filtering. IGV is a sequencing sequence of visualization software, through the BWA sequencing sequence comparison to generate Sam files, Sam software Sam file generated by Sam Bam file, IGV software through the Bam file visualization can be seen with the hg19 bp A comparison was made of SNPs. The experiment used in this article is targeted double-ended sequencing of breast cancer data (http://pan.baidu.com/s/ 1kVNlWQb), 15 samples were used for correlation analysis.
The following table is the location index method, Bcftools, IGV under the artificial search results (Table 2).
| Sample | Location index | Bcftools | IGV |
|---|---|---|---|
| Sample 1 | 65 | 38 | 35 |
| Sample 2 | 78 | 55 | 49 |
| Sample 3 | 52 | 41 | 38 |
| Sample 4 | 45 | 40 | 30 |
| Sample 5 | 79 | 45 | 39 |
| Sample 6 | 64 | 55 | 50 |
| Sample 7 | 51 | 42 | 38 |
| Sample 8 | 54 | 46 | 43 |
| Sample 9 | 46 | 40 | 37 |
| Sample 10 | 58 | 44 | 41 |
| Sample 11 | 68 | 54 | 46 |
| Sample 12 | 64 | 52 | 47 |
| Sample 13 | 52 | 45 | 40 |
| Sample 14 | 59 | 51 | 47 |
| Sample 15 | 43 | 37 | 32 |
Table 2: The algorithm is compared with other methods.
As can be seen from the table above, the proposed method can find more SNP points, and bcftools and IGV can find SNP points less, through the analysis of bcftools and IGV analysis process for the following reasons:
1. If there is a large number of inversion sequences in the original sequence, this will result in the reverse sequence cannot be Alignment to the corresponding gene location, subsequent mutation detection is difficult to be identified.
2. False-negative problems, there are many similar sequences in the DNA sequence, may lead to relative sequencing data to other locations, such as: breast cancer targeting sequencing gene CHEK2 exon, located in chr22: 29085073- 29085244 target Sequence, whereas sequencing sequences were located in chr16: 32369488-32369560. This will lead to a large number of data loss, causing false negative problems occurred in this experiment exon position in chr2, chr3, chr5, chr9, chr10, chr11, chr13, chr16, chr17, chr19, chr22 and BWA generated to Sam file, The chr1, chr4, chr6, chr15, chr18, and chrX can all be compared to the corresponding sequences, and these are not the original sequence, resulting in a large number of false positives or false negative phenomena exist, will lose a lot SNP points.
Conclusion
Through the traditional detection method, there are a large number of sequences cannot be compared to the DNA sequence, through the proposed positional index based on the SNP algorithm can directly compare the original sequencing sequence to the target sequence and Found more SNP points, which greatly improved the discovery of key SNP points for the relevant diagnosis to provide more data support.
Acknowledgements
This paper was supported by the National Nature Science Foundation of China under contract number 61271259 and 61601070, the Chongqing Nature Science Foundation under contract number CTSC2011jjA40006, CSTC2010BB2415 and CSTC2016jcyjA0455, the Research Project of Chongqing Education Commission under contract number KJ120501, KJ12050, KJ1600411and KJ110530.
References
- Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet 2006; 7: 61-80.
- Kim BC. SNP @ Promoter: a database of human SNPs (single nucleotide polymorphisms) within the putative promoter regions. BMC Bioinform 2008; 9: 279-282.
- Yang JO, Kim WY, Bhak J. ssSNPTarget: genome-wide splice-site Single Nucleotide Polymorphism database. Hum Mutat 2009; 30: E1010-1020.
- Hariharan M, Scaria V, Brahmachari SK. dbSMR: a novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinform 2009; 10: 108.
- Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 2008; 18: 1851-1858.
- Li R, Li Y, Fang X, Yang H, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res 2009; 19: 1124-1132.
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics 2008; 24: 713-714.
- Goya R, Sun MG, Morin RD, Leung G, Ha G. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics 2010; 26: 730-736.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J. The Sequence alignment/map format and SAMtools. Bioinformatics 2009; 25: 2078-2079.
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K. The genome analysis toolkit: a mapreduce framework for analysing next-generation DNA sequencing data. Genome Res 2010; 20: 1297-1303.
- Fenzhen C, Ling L. Comparing of four common biological sequence alignment tools. Chin J Bioinform 2016;14: 56-60.
- Tung N, Battelli C, Allen B, Kaldate R. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015; 121: 25-33.
- Meindl A, Hellebrand H, Wiek C. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Gene 2010; 42: 410-414.
- Tommiska J, Seal S, Renwick A, Barfoot R, Baskcomb L. Evaluation of RAD50 in familial breast cancer predisposition. Int J Cancer 2006; 118: 2911-2916.
- Harkness EF, Barrow E, Newton K, Green K, Clancy T. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet 2015; 52: 553-556.
- Marzec KA, Martino-Echarri E. BARD1 splice variants display mislocalization in breast cancer cells and can alter the apoptotic response to cisplatin. Cancer Lett 2016; 381: 149-155.
- Cremona CA, Behrens A. ATM signalling and cancer. Oncogene 2014; 33: 3351-3360.
- Ataei-Kachouei M, Nadaf J, Akbari MT, Atri M, Majewski J. Double heterozygosity of BRCA2 and STK11 in familial breast cancer detected by exome sequencing. Iran J Public Health 2015; 44: 1348-1352.
- Maggi LB, Weber JD. Targeting PTEN-defined breast cancers with a one-two punch. Breast Cancer Res 2015; 17: 51.
- Da SA, Feldman L. Epigenetic modifications, chromatin distribution and TP53 transcription in a model of breast cancer progression. J Cell Biochem 2015; 116: 533-541.
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007; 39: 165-167.
- Van Rs, Vogelaar T, Carneiro F. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Gene 2015; 52: 361-374.
- Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN. Germline mutations in the BRIP1, BARD1, PALB2, and NBN Genes in women with ovarian cancer. J Natl Cancer Inst 2015; 107.
- Wang N, Ding H, Liu C. A novel recurrent CHEK2 Y390C mutation identified in high-risk Chinese breast cancer patients impairs its activity and is associated with increased breast cancer risk. Oncogene 2015; 34: 5198-5205.
- Volkdraper L, Hall K, Griggs C. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res 2014; 74: 5421-5434.
- Pham TT, Angus SP, Johnson GL. MAP3K1: genomic alterations in cancer and function in promoting cell survival or apoptosis. Genes Cancer 2013; 4: 419-426.
- Hunter Dj, Kraft P, Jacobs KB. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nature Gene 2007; 39: 870-874.
- Jones JO, Chin SF, Wong-Taylor LA, Leaford D, Ponder BA. TOX3 mutations in breast cancer. PLoS One 2013; 8: e74102.
- Abaildayev AO, Mukushkina DD, Neupokoyeva AS. Association rs909116 in gene LSP1 with breast cancer in Kazakhstan populations. J Biotechnol 2016; 231: S93-S94.
- Stahl P, Seeschaaf C, Lebok P, Kutup A, Bockhorn M. Heterogeneity of amplification of HER2, EGFR, CCND1 and MYC in gastric cancer. BMC Gastroenterol 2015; 15: 7.