- Journal of RNA and Genomics (2006) Research Article
3'-Poly(A) tail enhances siRNA activity against exogenous reporter genes in MCF-7 cells
| Jie Li, Guang Yang, Shaohua Li, Guojun Cao, Qiang Zhao, Xuemei Liu, Ming Fan, Beifen Shen, Ningsheng Shao* Beijing Institute of Basic Medical Sciences, 27 Taiping Road, Haidian District, Beijing 100850, China |
| Corresponding Author: Ningsheng Shao, Email: shaons@yahoo.com, Tel: +010 68163140, Fax: +010 68163140 |
| Received 06 May 2006; Revised 01 July 2006; Accepted 10 July 2006; Available online 17 July 2006; Published 28 July 2006 |
Abstract
RNA interference (RNAi) is a powerful tool in the study of gene function. We added poly(A) tails to the 3' ends of siRNA antisense strands by in vitro transcription, and investigated the silencing effects of poly (A)- tailed siRNAs on enhanced green fluorescence protein (EGFP) and red fluorescence protein (RFP) genes. The results of this study showed that siRNAs with single-stranded 3’-poly(A) tails at antisense strands had noticeably stronger silencing effect on exogenous reporter genes than their corresponding parental forms. The enhanced silencing effect appears to be related to the length of poly(A) but was non siRNA sequencespecific. Furthermore, our results demonstrate that weakly-activated PKR and reduced stability of mRNAs of exogenous reporter genes in vivo may be the possible mechanisms of this non-specific enhanced silencing effect. Our findings are likely to be of value in designing siRNAs with enhanced activity.
KEYWORDS |
| siRNA, poly(A) tail, non-specific, enhanced gene silencing |
INTRODUCTION |
| RNA interference (RNAi) is the process by which dsRNA silences gene expression, either by inducing the sequencespecific degradation of complementary mRNA or by inhibiting translation. RNAi was initially defined in the worm Caenorhabditis elegans by Fire et.al (Fire et al, 1998), and we now know it is conserved from fungi to mammals. The RNAi pathway has many cellular functions, including protecting cells against viruses and repetitive DNA elements, directing the assembly of heterochromatin, and regulating cellular development, growth, differentiation, and apoptosis. A simplified model for RNAi pathway involves two steps. In the first step, long double-stranded RNA or microRNA primary transcripts are converted to segments of short interfering RNA (siRNA), which are 21-23 bp long, by the RNase IIIlike protein Dicer (Bernstein et al, 2001), leaving 5´- phosphate 3´-hydroxy termini and 2 nt 3´ overhangs. In the second step, these products are incorporated into the RNAinduced silencing complex (RISC) (Hammond, 2005). The siRNA is unwound in a strand-specific manner during RISC formation. Gene silencing results from translational inhibition or degradation of target mRNA, depending upon the siRNA/mRNA duplex complementarity. |
| Long dsRNAs (>30 bp) cannot be used in mammals with provoking the antiviral/interferon response (Williams, 1999). It was shown that siRNAs with 21-23 bp which can be synthesized chemically or transcribed in vitro, could bypass the interferon pathway (Elbashir et al, 2001; Paddison et al, 2002). However, recent reports suggest some otherwise (Sledz et al, 2003; Bridge et al, 2003). Adding dTdT or UU to the 3´ end of siRNAs may improve silencing efficiency based the finding that the PAZ domain can interact with the 3´ end of single-stranded RNA. The PAZ domain, which is found in Dicer family proteins and Argonaute proteins, can bind 3´ overhangs of siRNAs, possibly anchoring the 3´ end of guide RNA within the silencing effector complex (Lingel et al, 2003; Song et al, 2003; Yan et al, 2003). However, dTdT and UU are not the only 3´ overhangs of siRNAs, and the effects of alternative 3´ overhang sequences on the efficiency and specificity of RNAi are unclear. In the present study, we investigated the effects on the activity of siRNAs when a single-stranded poly(A) was added to the 3´ end of the antisense strand. |
MATERIALS AND METHODS |
| Synthesis and purification of RNA |
| DNA oligonucleotides with T7 primers were annealed to produce dsDNA fragments. All RNAs were synthesized by in vitro transcription, purified using denaturing polyacrylamide gel (Bevilacqua and Cech, 1996), and treated with calf intestinal alkaline phosphatase (Promega) as described previously (Kim et al, 2004). The ssRNAs were annealed to form dsRNAs in annealing buffer (50 mM Tris, pH 7.5, 100 mM NaCl in DEPC-treated water). The RNAs used in these studies were as follows:- |
| EGFP siRNA: |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-AACUUCAGGGUCAGCUUGCCG-3' |
| EGFP siRNA-18A (gsi18A): |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-ACUUCAGGGUCAGCUUGCCGA18-3' |
| EGFP siRNA-36A (gsi36A): |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-AACUUCAGGGUCAGCUUGCCGA36-3' |
| EGFP siRNA-60A (gsi60A): |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-AACUUCAGGGUCAGCUUGCCGA60-3' |
| EGFP siRNA-60U (gsi60U): |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-AACUUCAGGGUCAGCUUGCCGU60-3' |
| EGFP siRNA-60N (gsi60N,N-negative): |
| 5'-CAAGCUGACCCUGAAGUUCAU-3' |
| 5'-AACUUCAGGGUCAGCUUGCCGGGUAACGCCAG GGUUUUCCGUCCUUUGUCGAUACUGGCUAAAUC CACUGUGAUAUCUAAU-3' |
| Negative control siRNA (nsi): |
| 5'-UUCUCCGAACGUGUCACGUUG-3' |
| 5'-ACGUGACACGUUCGGAGAAUG-3' |
| Negative siRNA -18A (nsi18A): |
| 5'-UUCUCCGAACGUGUCACGUUG-3' |
| 5'-ACGUGACACGUUCGGAGAAUG A18-3' |
| Negative control siRNA -36A (nsi36A): |
| 5'-UUCUCCGAACGUGUCACGUUG-3' |
| 5'-ACGUGACACGUUCGGAGAAUG A36-3' |
| Negative control siRNA -60A (nsi60A): |
| 5'-UUCUCCGAACGUGUCACGUUG-3' |
| 5'-ACGUGACACGUUCGGAGAAUG A60-3' |
| RFP siRNA (rsi): |
| 5'-AGUUCCAGUACGGCUCCAAGG-3' |
| 5'-UUGGAGCCGUACUGGAACUUG-3' |
| RFP siRNA-18A (rsi18A): |
| 5'-AGUUCCAGUACGGCUCCAAGG-3' |
| 5'-UUGGAGCCGUACUGGAACUUG A18-3' |
| RFP siRNA-36A (rsi36A): |
| 5'-AGUUCCAGUACGGCUCCAAGG-3' |
| 5'-UUGGAGCCGUACUGGAACUUG A36-3' |
| RFP siRNA-60A (rsi60A): |
| 5'-AGUUCCAGUACGGCUCCAAGG-3' |
| 5'-UUGGAGCCGUACUGGAACUUG A60-3' |
| 10A: 5'-A10-3' |
| 20A: 5'-A20-3' |
| 30A: 5'-A30-3' |
| 40A: 5'-A40-3' |
| 50A: 5'-A50-3' |
| 60A: 5'-A60-3' |
| 60U: 5'-U60-3' |
| 60N (N is negative control): 5'-GGTAACGCCAGGGTTT TCCGTCCTTTGTCGATACTGGCTAAATCCACTGTG ATATCTAAT-3' |
| Transfection and silencing assay |
| All transfections were carried out using lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). MCF-7 cells (90% confluent) were transfected in 24-well plates. A reporter gene vector (200 ng) and 40-nM RNA were co-transfected, and the ratio of lipofectamine:DNA+RNA (μl:μg) was 2:1. Reporter gene expression was visualized by fluorescence microscopy 48 hr after transfection. The reporter gene expression vectors pEGFP-N2 (EGFP) and pdsRED2-N1 (RFP) were purchased from Clontech. |
| RT-PCR analysis of mRNA and DNA |
| Total RNA and DNA were isolated from cells using TRIzol® reagent (Invitrogen). cDNAs were generated using M-MLV Reverse Transcriptase (Promega) and oligo(dT)15 primer (Promega) according to the manufacturer’s instructions, typically using 500 ng of total RNA per reaction. The polymerase chain reaction (PCR) was performed on 2 μl cDNA in 50 μl of final volume. Samples were analyzed by electrophoresis on 1.5% (w/v) agarose gel and visualized by ethidium bromide staining. The results were normalized with R-actin internal control. The various primers are below: |
| The EGFP primers: |
| 5S-GCAAGCTGACCCTGAAGTTCATC-3S (forward) |
| 5S-TCACCTTGATGCCGTTCTTCTG-3S (reverse) |
| The RFP primers: |
| 5S-CGAGGGCTTCAAGTGGGA-3S (forward) |
| 5S-CTACAGGAACAGGTGGTGGC-3S (reverse) |
| The β-actin primers: |
| 5S-CATCTCTTGCTCGAAGTCCA-3S (forward) |
| 5S-ATCATGTTTGAGACCTTCAACA-3S (reverse). |
| Bacterial expression and purification of GST-PKR |
| The PKR expression plasmid, pEGST-PKR/TPP, was a gift from Dr Takayasu Date at Kanazawa Medical University. GST-PKR was expressed in Eschierichia coli BL21 and purified as described previously (Matsui et al, 2001). |
| In vitro PKR kinase assay |
| GST-PKR (2 Ug) was dephosphorylated with 400 U of T- PPase (Upstate, Charlottesville,VA) at 37°C for 30 min, and the reaction was quenched with 2 mM sodium orthovanadate (Sigma). A 10 Ul mixture containing 0.5 Ug of treated GST-PKR, 20 UM ATP, 10 UCi of [X-32P]ATP, the designated concentrations of RNAs and buffer (50 mM Tris-HCl, 2 mM MgCl2, 50 mM KCl, pH 7.5) was incubated for 30 min at 23°C. Reactions were quenched using SDS sample buffer. Phosphorylated proteins were separated via SDS/PAGE and visualized by autoradiography. |
| Cell lysis and immunoblotting |
| Cells were washed with PBS (25°C) and then lysed over 20 min in cold lysis buffer (50 mM Tris-HCl; pH 7.4; 1% (v/v) NP-40; 0.25% (w/v) sodium deoxycholate; 150 mM NaCl; 1 mM each EDTA, PMSF, Na3VO4, and NaF; 1 μg/ml each aprotinin, leupeptin, and pepstatin). Extracts were clarified by centrifugation (10,000 × g) for 20 min at 4°C. Protein concentrations were determined using the Bradford assay. Proteins were separated using 10% (w/v) SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was blocked in a solution of TBS containing 5% (w/v) non-fat dry milk and 0.05% (v/v) Tween-20 (TBST-MLK) for 30 min at 25°C with constant agitation, incubated with primary antibodies per the manufacturer’s instructions for 2 hr at 25°C, washed with TBST, and incubated for 1 hr with a goat anti-rabbit HRP-conjugated IgG (1:5000 dilution). After three additional washes, enhanced chemiluminescence (ECL) was performed according to manufacturer’s protocol (Amersham). The anti-phospho-PKR (Thr446, rabbit polyclonal IgG) was purchased from Upstate, and the polyclonal antibodies against R-actin were obtained from Santa Cruz. |
| Preparation of 32P-labled mRNA fragments |
| A 262-base fragment of EGFP mRNA (bases 46-308) was prepared by in vitro transcription from DNA templates containing T7 RNA polymerase promoter. The DNA templates were prepared by PCR amplification using the following primers: |
| 5´-GCATGCTAATACGACTCACTATAGGGCACATC GCTCAGACACCA-3´ (forward) |
| 5´-GAGCACAGGGTACTTTATTGA-3´(reverse) |
| Poly(A) tail was added to the 3´ end of the siRNAs using poly(A) polymerase (TaKaRa) as per the manufacturer’s protocol. The 5´ end of poly(A) mRNA was labeled using T4 polynucleotide kinase and [X-32P]ATP (MEGALABEL™ Kit, TaKaRa). |
| In vitro mRNA degradation |
| In vitro mRNA degradation assay was modified according to the method described previously (Ross and Kobs, 1986; Tuschl et al, 1999). Briefly, MCF-7 cells were transfected with different tiny RNAs (40 nM). After 12 hr, cellular extracts from were prepared by homogenization in lowsalt buffer (10 mM Tris-HCl pH 7.6, 1 mM magnesium acetate, 1.5 mM potassium acetate, 2 mM dithiothreitol, 0.1 mM PMSF, 1 μg/ml each aprotinin, leupeptin, pepstatin). mRNA was allowed to decay at 37°C for various lengths of time. Reactions took place in 20 μl volumes containing 0.2 A260 U of cellular extracts, 10 nM tiny RNAs [siRNA, siRNA-poly(A) or poly(A)], and 50 pM mRNA (containing equal amounts of 5S-32P-labled mRNA) in buffer [10 mM Tris-HCl pH 7.6, 2 mM magnesium acetate, 1 mM potassium acetate, 2 mM dithiothreitol, 10 mM creatine phosphate, 0.1 U creatine phosphokinase, 1 mM ATP, 0.4 mM GTP, 0.1 mM spermine, and 2 U of RNasin (Promega)]. At each time point, RNA was extracted and processed via electrophoresis in 4% (w/v) polyacrylamide 7 M urea gels (Bernstein et al, 1989). |
| Measurement of mRNA stability in vivo |
| The EGFP reporter gene was transfected in MCF-7 cells for 12 hr. Cells were then transfected with groups of RNA (40 nM), and Actinomycin D (ActD, 10 μg/ml) was added during RNA treatment as described previously (Jing et al, 2005). Total RNA was isolated at different time points after RNA treatment. The stability of EGFP mRNA was determined via quantitative PCR using an Applied Biosystems Prism 7000, SYBR Green PCR MasterMix (QiaGen, TIANGEN), and specific primers (listed below) to detect EGFP and GAPDH. Target gene expression was normalized using a standard curve, and data were analyzed using Applied Biosystems Prism software and the \\Ct method (Vandesompele et al, 2002). Each sample was analyzed in duplicate. |
| The EGFP primers (for qPCR): |
| 5S-CGACGGCAACTACAAGAC-3S (forward) |
| 5S-TAGTTGTACTCCAGCTTGTGC-3S (reverse) |
| The GAPDH primers (for qPCR): |
| 5S-GGGTGTGAACCATGAGAAGT-3S (forward) |
| 5S-GGCATGGACTGTGGTCATGA-3S (reverse) |
RESULTS |
| siRNAs with poly(A) tails enhance the silencing of exogenous reporter genes |
| siRNA and siRNA-poly(A) were prepared with 18A, 36A or 60A tails on the 3´ ends of their antisense strands for EGFP and RFP. These compounds were designated gsi, gsi18A, gsi36A, gsi60A, rsi, rsi18A, rsi36A, and rsi60A, respectively. The silencing efficiency of the prepared siRNAs on EGFP and RFP were firstly tested. In 24-well plates, MCF-7 cells were transfected with siRNA (40 nM) and reporter plasmid (200 ng). Under the fluorescence microscope, it was apparent that gsi could silence EGFP but had no effect on RFP, while rsi could silence RFP but had no effect on EGFP. Negative siRNA (nsi, a randomized non-specific control siRNA) had no effect on both EGFP and RFP (Figure 1A). Then, the silencing effect of gsi and gsi-poly(A) on EGFP were compared. Interestingly, the results showed that EGFP expression was silenced more efficiently by gsi-poly(A) than by gsi alone. Changes in EGFP mRNA levels displayed similar silencing effects (Figure 1B). The silencing activity of RFP siRNA and rsi-poly(A) on exogenous RFP expression was also investigated, and results indicated that RFP expression was inhibited by rsi and to a greater extent than by rsi-poly(A) (Figure 1C). Furthermore, the extent of inhibition appeared directly proportional to the length of the poly(A) tail. Non-sequence-specific |
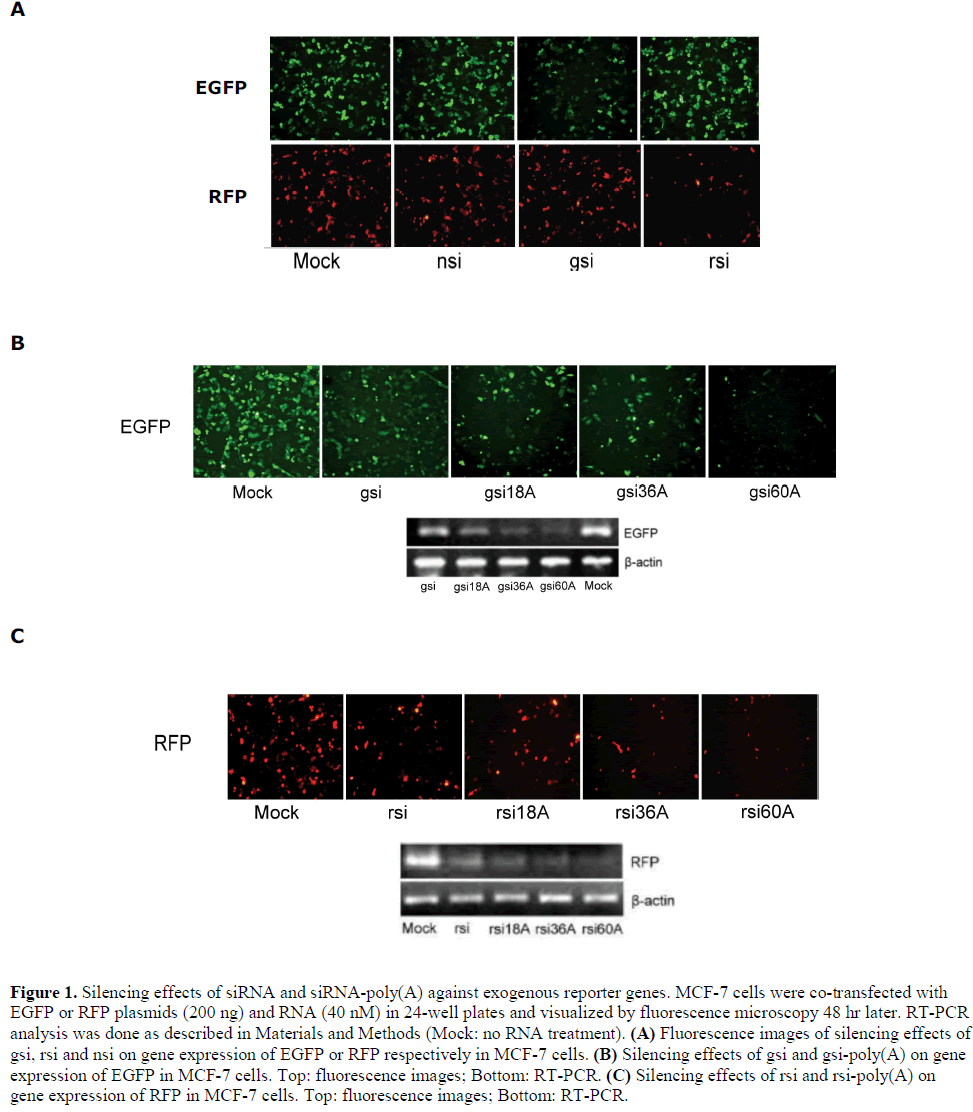 |
| Non-sequence-specific effects of siRNA-poly(A) on exogenous reporter genes |
| A specific silencing effect is an important characteristic of siRNA. To test the specificity of siRNA-poly(A), we co-transfected MCF-7 cells with pEGFP-N2 and pdsRED2- N1 expression plasmids and gsi60A. The results showed that gsi60A inhibited the expression of both EGFP and RFP when compared with the MOCK group, but gsi inhibited EGFP expression only (Figure 2A). DNA was extracted from cells and assayed using semi-quantitative PCR for reporter gene plasmids. However, no obvious differences were discernable among the groups (data not shown). The inhibition by gsi60A was probably not a result of differences in transfection efficiency. While comparing gsi sense and antisense sequences with the RFP mRNA sequence, we did not observe high homology or “seed regions” that are characteristic of microRNAs. The inhibition of RFP expression by gsi60A was apparently a non-specific effect. The strength of the inhibition was related directly to the length of poly(A) tail (Figure 2B). We obtained similar results with rsi-poly(A) and nsipoly( A) (data not shown). The strength of the inhibition was related directly to the length of poly(A) tail. We suggest that the inhibition of exogenous EGFP and RFP expressions was non-specific. |
| Poly(A) tail causes non-specific inhibition |
| Since gsi60A exhibited improved silencing efficiency, the dependence of non-specific inhibition on poly(A) sequence was investigated. The polynucleotide chains 60U and 60N (N, randomized nucleotides) were substituted for 60A. We found that gsi60U, gsi60N, and gsi all inhibited EGFP expression but had no effect on RFP gene expression (Figure 3A). Interestingly, 60A itself conferred a non-specific silencing effect on exogenous EGFP gene expression; 60U, poly(G) and poly(C) showed no such effect (Figure 3B). We then used poly(A)s of various lengths (10A, 20A, 30A, 40A, 50A, and 60A) in a silencing activity assay. The results demonstrated that single strands of poly(A) that were longer than 50 nt could silence EGFP expression non-specifically (Figure 3C). We further compared the silencing effect of gsipoly( A) and nsi-poly(A) on exogenous EGFP. The results showed that with the same length of poly(A) tails, gsipoly( A) had stronger silencing activity than nsi-poly(A) (Figure 3D), which suggested that both of the EGFP specific-siRNA and the poly(A) contributed to the silencing effect of gsi-poly(A) on EGFP. |
| siRNA-poly(A) can activate double-stranded RNAactivated protein kinase (PKR) |
| Interferon response is one of reasons for the nonspecific silencing effects of siRNA. Activation of PKR is the main event involved in this pathway. PKR is a double-stranded RNA-activated protein kinase. PKR uses its two Nterminal dsRNA binding domains as sensors to assist in inhibiting translation. It is well known that dsRNA strands that are longer than 30 bp can activate PKR. However, previous reports have demonstrated that short dsRNA, including 21 bp siRNA (Sledz et al, 2003; Bridge et al, 2003; Kim et al, 2004) and 16 bp dsRNA stems flanked by 10-15 nt single-stranded tails, could also activate PKR (Zheng and Bevilacqua, 2004). For these reasons, the effects of siRNA-poly(A) on PKR were investigated. |
| siRNA-poly(A)s activated PKR during an incubation with dephosphorylated PKR in the presence of [X-32P]ATP, and the strength of activation of PKR was directly related to the length of poly(A) tail. However, siRNA or poly(A) of 60nt individually could not activate PKR in vitro (Figure 4, A and B). The activating ability of gsi60A, nsi60A, gsi60U and gsi60N on dephosphorylated PKR were also compared in vitro, and among these, only siRNAs with poly(A)s (gsi60A and nsi60A) could activate PKR (Figure 4C). |
| Activation of PKR was observed in MCF-7 cells following transfection with gsi60A and nsi60A, but the effects were weaker than those of polyI:C (polyinosinic acidpolycytidylic acid, a dsRNA analog). gsi and nsi did not activate PKR. Interestingly, 60A activated PKR weakly ex vivo (Figure 4D). |
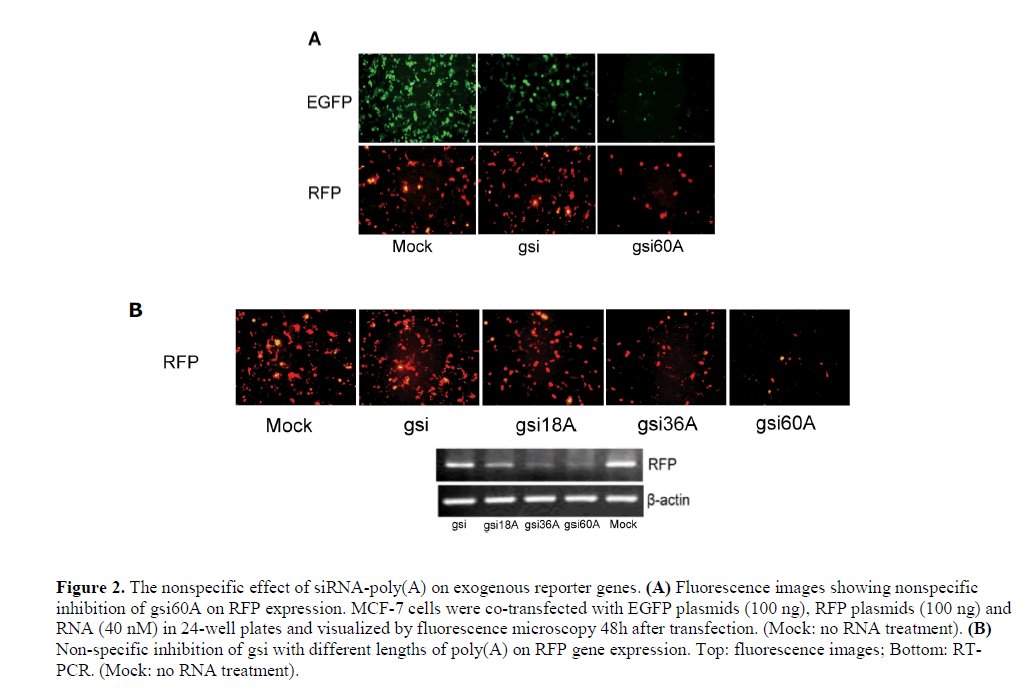 |
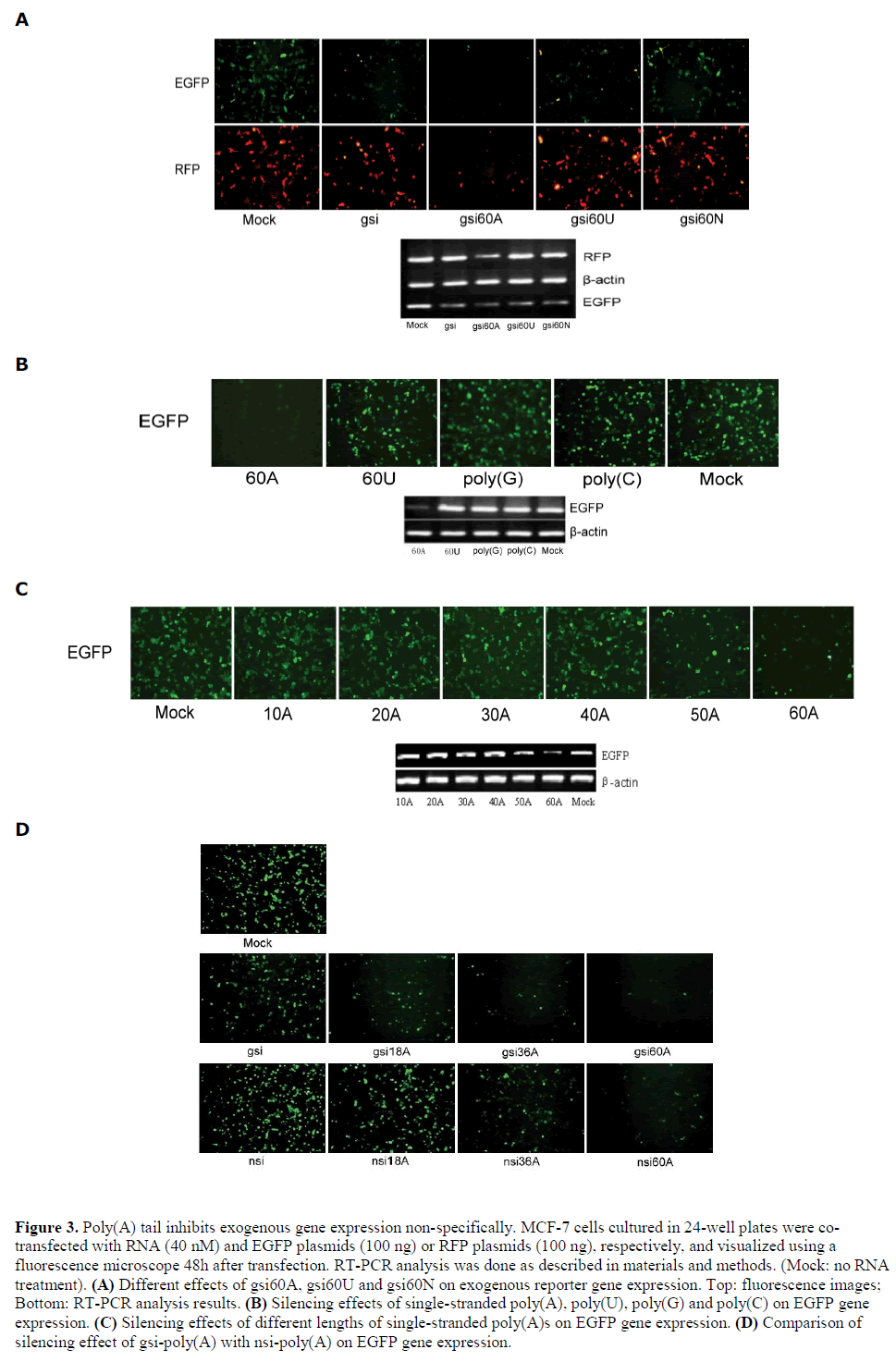 |
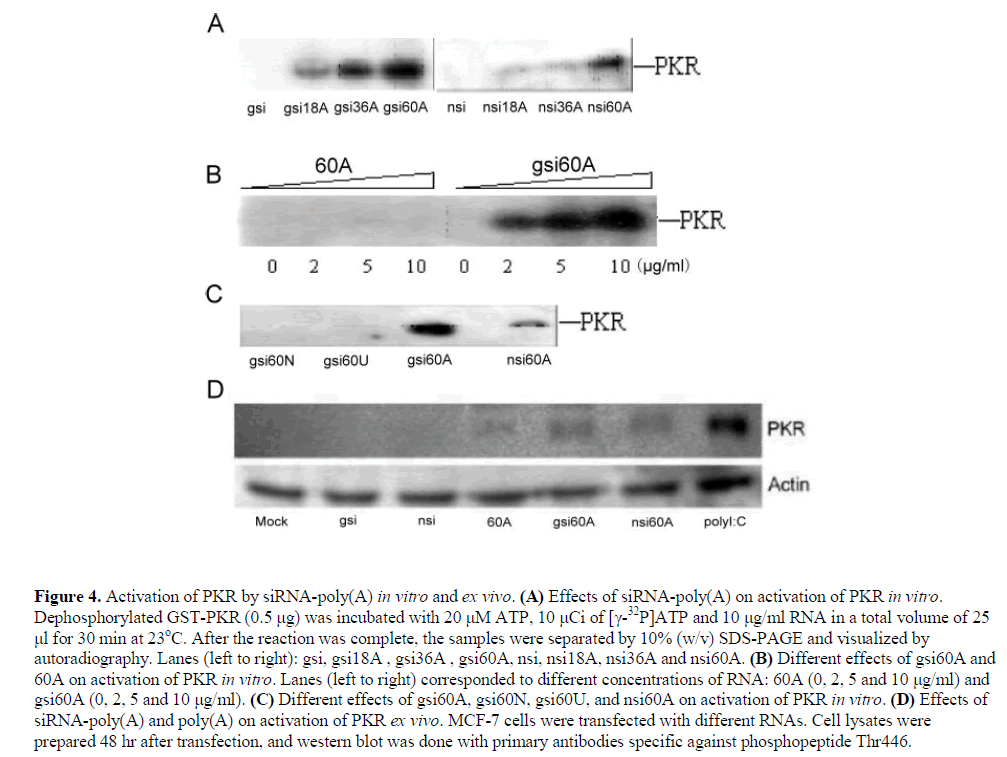 |
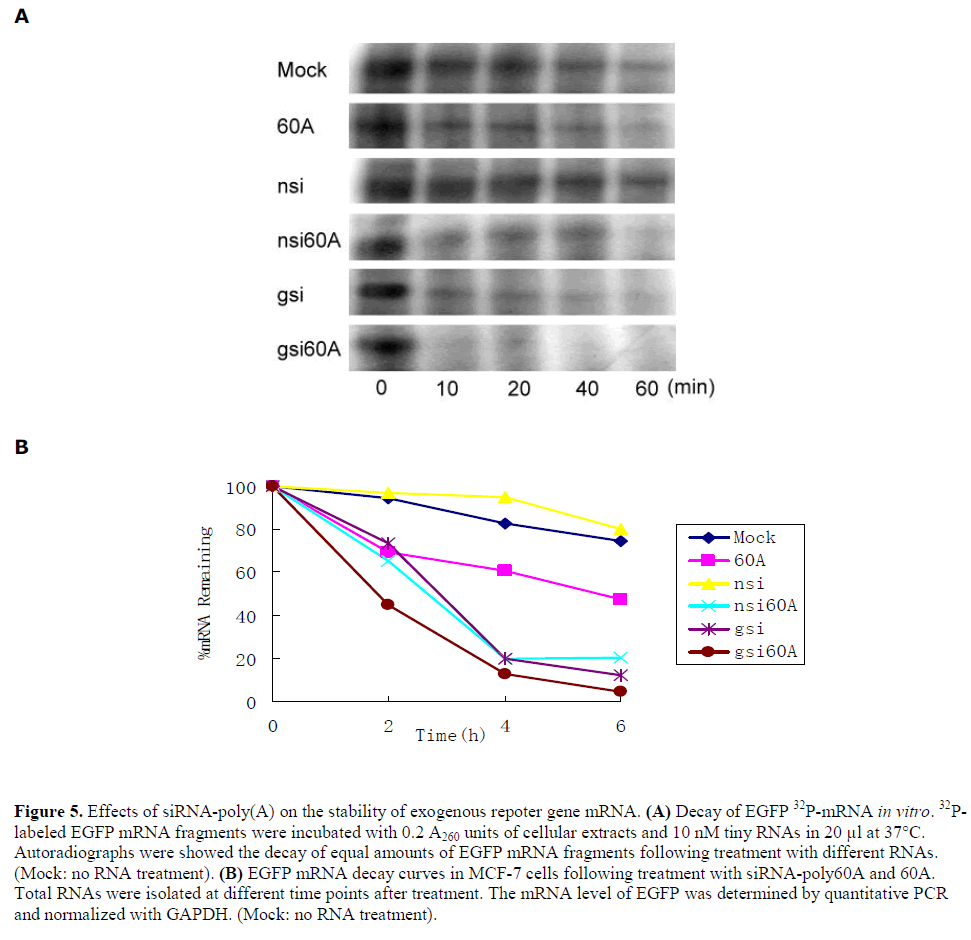
| Effects of siRNA-poly(A) on the stability of exogenous repoter gene mRNA |
| The effect of siRNA-poly(A) and poly60A on the stability of exogenous gene mRNA was investigated. First, mRNA decay was assessed by incubation of 5´-32P-labled EGFP mRNA fragments with MCF-7 cell extracts and different RNAs in vitro. The results demonstrated an enhancement of mRNA degradation rates by nsi60A, gsi60A and poly60A, but not nsi. In other words, siRNA-poly(A), including poly60A, reduced exogenous gene mRNA stability in this cell-free decay system. gsi caused rapid decay of EGFP mRNA because of its RNAi effect (Figure 5A). Then in vivo mRNA decay assay was done with MCF-7 cells. After treatment, EGFP mRNA levels were analyzed by real-time RT-PCR at different times. The stability of EGFP mRNA in cells treated with gsi60A, nsi60A or 60A was lower than in cells treated with nsi. Gsi also silenced EGFP expression effectively because of its RNAi effect (Figure 5B). |
DISCUSSION |
| In this study, it was demonstrated for the first time that single-stranded poly(A), following addition to the 3´ end of an siRNA antisense strand could enhanced the silencing effect of the siRNA on exogenous reporter gene expression. This enhancement was independent of the siRNA sequence and related directly to the length of the poly(A) strand. Our results showed that siRNA-poly(A) could activate PKR both in vitro and ex vivo. Although siRNA-poly(A) was a weaker activator of PKR than polyI:C, inhibition of translation by activated PKR could not be ruled out. Persengiev et al suggested that siRNAs affect nonspecific pathways that were overlapping but not identical to those regulated by interferon and polyI:C (Persengiev et al, 2004). That is, siRNAs do not trigger a true interferon response. In fact, this represented a possible mechanism of the non-specific enhanced RNA-silencing activity of siRNA-poly(A). Interestingly, single-stranded poly60A did not activate PKR in vitro but it could make PKR phosphorylation weakly ex vivo, representing a topic for further investigation. SiRNA-poly(A)s, including poly60A, also reduced the mRNA stability of the exogenous reporter EGFP gene, possibly representing another reason for their non-specific enhanced-silencing activity. Considering the feasibility of both explanations, the mechanism of action remains undefined. |
| Bernstein et al have suggested that the poly(A)-poly(A) binding protein complex influences mRNA stability in vitro (Bernstein et al, 1989). The transfected siRNApoly( A) and poly60A might compete with poly(A) binding protein (PABP) to influence the stability of mRNAs of exogenous genes. In order to verify this, siRNA-poly(A)s targeting EGFP were co-transfected with GSTPABP1plasmid (Lee and Bedford, 2002) in MCF-7 cells. We found that the silencing effect of siRNA-poly(A) on EGFP could not be reduced. RT-PCR results showed that exogenous PABP1 mRNA level was decreased to normal by siRNA-poly(A)s, but endogenous PABP1 mRNA level was not affected (data not shown). We also compared the effects of siRNA-poly(A)s on exogenous EGFP with stable expressed EGFP transgene. Surprisingly, the EGFP expression in stable transgene cells was not affected (data not shown). All this suggested that poly (A) tail had few effects on endogenous genes, but this needs to be further confirmed in the future. |
 |
| Previously, Kawasaki and Taira added a poly(A) sequence to the 3S end of a hammerhead ribozyme, leading to extremely efficient cleavage of the target mRNA (Kawasaki and Taira, 2002). The poly(A) sequence of the ribozyme interacted with eIF4A1, an RNA helicase, and cleaved the target mRNA regardless its secondary structure. Most recently, some interesting findings pointed to a relationship between poly(A) tails and the RNA interference pathway. When AGO2 or Dicer2 (essential factor for RNAi) in Drosophila was depleted, the reporter transgene EGFP protein levels rose and the mRNA poly(A) tails were shortened (Siomi et al, 2005). Humphreys et al reported that microRNAs controled translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function (Humphreys et al, 2005). Wu et al found that microRNAs expedite poly(A) removal and lead to rapid mRNA decay (Wu et al, 2006).These results above suggested possible links between gene silencing and poly(A) tails beyond our traditional understandings. |
| We now know that many non-translatable mRNA-like RNA transcripts are present in cells (Erdmann et al, 2000). They are polyadenylated, spliced and lack long ORFs. Many reports have suggested that these RNAs play a variety of structural, informational, catalytic and regulatory roles in the cell. However, only a few short mRNA-like RNAs (< 200bp) have been reported in human cells. As siRNA-poly(A) in this study looks like a short mRNA and non-specifically inhibits exogenous gene expression, we guessed the possibility that short mRNAlike RNA may have some important biological roles especially in mammalian cells. |
| In this paper, EGFP and RFP were used as exogenous reporter genes, but we believe that our results provide a functional connection between poly(A) sequence, siRNA and gene silencing. Our findings may be useful in siRNA designing especially when the targets are exogenous genes or against other invading virus RNAs. |
CONCLUSIONS |
| Single-stranded poly(A) tails on the 3' ends of siRNAs antisense strands have stronger silencing effects on exogenous genes than their corresponding parental forms. The enhanced silencing effect is related to the length of poly(A) but is non-specific. The nonspecific enhanced silencing effect may be related to the weak activation of PKR and the reduced stability of exogenous genes mRNAs. These results contribute the design of siRNAs and suggest the possible application of siRNA-poly(A) on cell defenses against exogenous genes or virus. |
ACKNOWLEDGMENTS |
| We thank Dr Takayasu Date and Tadashi Matsui for the gift of GST-PKR expression plasmid, and Dr Bedford for the gift of GST-PABP1 expression plasmid, and the other members of the Shao laboratory, namely Zhaohui Liu, Liucun Chen, Weiguo Sun, Jie Dong and Shuyun Liu, for many helpful suggestions. We thank Dr Benjamin M Johnson and Dr Tianjian Yang for their kind help in linguistic revision of this paper. This work was supported by the National “973” Program of China (Program No. 2005CB724600) and the Natural Science Foundation of China (Project No. 30470390, 30300067). |
STATEMENT OF COMPETING INTERESTS |
| Ningsheng Shao has two pending patents on the methods of this paper. |
LIST OF ABBREVIATIONS |
| siRNA-poly(A): poly(A) tailed siRNA |
| gsi: EGFP siRNA |
| gsi18A: EGFP siRNA-18A |
| gsi36A: EGFP siRNA-36A |
| gsi60A: EGFP siRNA-60A |
| rsi: RFP siRNA |
| rsi18A: RFP siRNA-18A |
| rsi36A: RFP siRNA-36A |
| rsi60A: RFP siRNA-60A |
| nsi: Negative siRNA |
| nsi18A: Negative siRNA -18A |
| nsi36A: Negative control siRNA -36A |
| nsi60A: Negative control siRNA -60A |
| PKR: Double-stranded RNA-activated protein kinase |
| polyI:C : Polyinosinic acid:polycytidilic acid |
| PABP: Poly(A) binding protein |
SHORT COPYRIGHT STATEMENT |
| This is an open access article, published under the terms of the Licence for Users available at http://www.libpubmedia.co.uk/ RNAiJ/LicenceForUsers.pdf. This licence permits noncommercial use, distribution and reproduction of the article, provided the original work is appropriately acknowledged with correct citation details. |
References |
|