- Journal of RNA and Genomics (2005) Review Article
Vector-based siRNA delivery strategies for high-throughput screening of novel target genes
| Meihong Chen1, Quan Du2, Hong-Yan Zhang2, Claes Wahlestedt2,3 and Zicai Liang2* 1Chinese Human Genome Center North, Beijing, 100176, China and Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC), National Laboratory of Medical Molecular Biology, Beijing, 100005, P. R. China. 2Center for Genomics and Bioinformatics, Karolinska Institutet, 17177, Stockholm, Sweden. 3Department of Drug Discovery, Scripps Florida, Jupiter, FL 33458, USA |
| Corresponding Author: Zicai Liang, Email: Zicai.Liang@cgb.ki.se, Tel: +46 8 5248 6371, Fax: +46 8 323950 |
| Received 10 June 2005; Revised 16 June 2005; Accepted 19 June 2005, Available online 27 July 2005; Published 12 August 2005 |
Visit for more related articles at Journal of RNA and Genomics
Abstract
Application of siRNA in high-throughput fashion is still in its early phase although the principle has been established for three years. In this review, we outline the different vector-based siRNA delivery platforms as well as resources that are becoming available for high-throughput applications, and some initial outcomes of vector siRNA high-throughput screening efforts using vector encoded siRNA. It is expected that further improvement of the siRNA technology and availability of the siRNA resources will help to materialize the potential of siRNA for functional genomics and drug target validation.
Keywords |
| siRNA, RNAi, vectors, gene silencing, siRNA delivery |
Introduction |
| RNA interference (RNAi) achieved by double-stranded RNA (dsRNA) mediated sequence-specific breakdown of the mRNA (Fire et al, 1998; Montgomery and Fire, 1998; Elbashir et al, 2001a). It is an evolutionarily conserved phenomenon initially characterized in C. elegans and Drosophila, but also occurs in plants, fungi and mammals (Boshare and Labouesse, 2000). Mechanistic studies have indicated that the effector molecules of the RNAi process is not the long dsRNA itself, but a family of short doublestranded RNA fragments (19-21 base pairs in length) derived through cleavage of the long dsRNA by RNase III enzyme(s) (Elbashir et al, 2001b). The short doublestranded RNA fragments generated as such, as well as through chemical synthesis or vector-based expression, are collectively referred to as small interfering RNA (siRNA). The long dsRNA cannot be used in mammalian cells for gene silencing due to the fact that introduction of long dsRNA elicits a strong antiviral response that obscures any gene-specific silencing effect. Much of this response is caused by activation of the dsRNA-dependent protein kinase PKR, which phosphorylates and inactivates the translation initiation factor eIF2a (Proud, 1995). Introduction of siRNA into mammalian cells was found not to elicit the antiviral response to such an extreme level, but still can result in efficient gene knockdown (Sledz et al, 2003; Elbashir et al, 2001b). This makes siRNA widely applicable in various mammalian species. The mechanism and biological implications of RNAi are still not fully understood. |
| Methods for generating siRNAs |
| Short siRNA molecules can be prepared by direct chemical synthesis or transcription driven by RNA polymerase promoters. In term of high-throughput applications, vector- based strategies are favoured because such strategies enjoy the advantages of much lower cost and ability to regenerate. A synthetic siRNA library could be expensive for most researchers. The only large-scale synthetic siRNA library that was produced so far is the genome-wide siRNA collection made for Novartis by Qiagen (Hilden, Germany) and Dhamarcon (Lafayette, USA). Transfection efficiency of siRNA into cells depends on cell types and the RNAi effect of synthetic siRNA only sustain for a period of time. The advantage of the vector-based siRNA is the capability of removing those cells that are not transfected with the plasmids by selecting the transfected cells with antibiotic resistant genes. Virus vectors also enable the delivery of siRNA expression cassettes into cells with higher transfection efficiency, and in case of lentivirus and retrovirus, it is easy to make stable knockdown cells by integration into the genome. If one wants to use siRNA expression vectors as a combinatorial library, the viral vectors would be most suitable. In the current review, we will focus on the vector-based siRNA libraries and the methods to produce them. |
| siRNA production by pol III promoters |
| The use of pol III promoters in vectors for encoding siRNA was pioneered by a number of groups in early 2002 (Brummelkamp et al, 2002; Miyagishi and Taira, 2002; Sui et al, 2002; Xia et al, 2002; and Yu et al, 2002). The vectors contain a single pol III promoter followed by a segment of DNA that encodes a short hairpin RNA (shRNA) (Figure 1A). The shRNA closely resembles the structure of microRNAs and will be integrated into the RISC complex in the same way as the normal siRNA does. The backbones of these constructs were initially plasmids, but the grafting the shRNA expressing cassettes into different viral vectors such as lentivirus, retrovirus and even adenovirus also explored successfully to facilitate the transfection of cells that were hard to penetrate, such as primary cells and suspension cells (Li et al, 2003; Hosono et al, 2004; Devroe and Silver, 2004) |
| Another strategy of encoding siRNA that has gain more popularity is a vector that contains dual pol III promoters arranged in a convergent manner that drive the expression of two short complementary RNA strand from a single 19 bp DNA region (Figure 1B) (Chen et al, 2005; Zheng et al 2004). While being as effective as the shRNA encoding vectors, these dual-promoter (GeneBuster) vectors are first of all more robust for cloning both because of the lack of secondary structure in the siRNA coding region, and much lower level of DNA synthesis errors due to the shorter sequence, and they have tremendous advantages in constructing large scale siRNA libraries in a combinatorial way, as will be discussed below. |
| A third method is a technique using two tandem pol III promoters to drive the expression of siRNA from separate 19 mer DNA fragments (Figure 1C) (Lee et al, 2002). This method might not be widely used due to the fact that construction of such a plasmid is considerably more cumbersome than the other two methods. This method as well as the shRNA method, however, offers the possibility to integrate mismatches into the resulting siRNA, thus possibly enhance the efficacy of the siRNA or increase the frequency of effective siRNA. |
| H1 and U6 promoters from human and mouse are the most widely used pol III promoters in the siRNA vector construction. Other promoters that have been used include promoters of tRNAVal and tRNAMet (Oshima et al, 2003; Boden et al, 2003). All these pol III promoters are active in a large variety of cells, and appear to perform similarly in different species. This makes it very easy to use the same vector in different experimental systems. One interesting feature of the pol III promoters is that they can be easily transformed into inducible promoters (van de Wetering et al, 2003). This feature is very important for some functional studies and screening. |
| In addition to plasmid-based systems, PCR-derived siRNA expression cassettes based on the single-promoter system have been shown to efficiently suppress gene activities in transfected cells (Castanotto et al, 2002). |
| siRNA production by other promoters |
| Other promoters that have been used for siRNA production include T7 promoter and CMV promoter (Xia et al, 2002; Holle et al, 2004). Since T7 promoter does not work in mammalian cells, strategies employing T7 or similar promoters normally can only support siRNA production in test tubes, but not within cells. People have overcome this shortcoming by either pre-load the promoter with CMV promoter is a much stronger promoter compared to other pol III promoters, and this can allow more siRNA molecules to be transcribed from a given amount of DNA vector. But since CMV promoter is an RNA polymerase II (pol II) promoter, the resulting transcripts are normally capped at the 5’-end and tailed at the 3’-end with a long poly (A) sequence. It appears that such modifications are well tolerated as shown in the efficacy assays. Due to the potentially higher degree of flexibility in enabling different spatial and temporal expression patterns, these types of vector-based siRNA strategies might be much more useful for in vivo research purposes and for siRNA based genetherapy at a later stage although pol III based vectors seem to be able to satisfy most of the gene knock-down needs in cultured cells. |
| Other promoters that have been used for siRNA production include T7 promoter and CMV promoter (Xia et al, 2002; Holle et al, 2004). Since T7 promoter does not work in mammalian cells, strategies employing T7 or similar promoters normally can only support siRNA production in test tubes, but not within cells. People have overcome this shortcoming by either pre-load the promoter with CMV promoter is a much stronger promoter compared to other pol III promoters, and this can allow more siRNA molecules to be transcribed from a given amount of DNA vector. But since CMV promoter is an RNA polymerase II (pol II) promoter, the resulting transcripts are normally capped at the 5’-end and tailed at the 3’-end with a long poly (A) sequence. It appears that such modifications are well tolerated as shown in the efficacy assays. Due to the potentially higher degree of flexibility in enabling different spatial and temporal expression patterns, these types of vector-based siRNA strategies might be much more useful for in vivo research purposes and for siRNA based genetherapy at a later stage although pol III based vectors seem to be able to satisfy most of the gene knock-down needs in cultured cells. |
Construction of siRNA libraries |
| One of the critical initial steps in the high throughput siRNA application is the formation of a physical siRNA library, which many people also refer to as a siRNA library. |
| To generate a siRNA library, one critical factor to consider is the design of siRNA sequence. Since the first guideline for siRNA design was published by Tuschl and colleagues (Elbashir et al, 2001b) several studies of currently available information for effective and non-effective siRNA have been carried out, and these have resulted in different siRNA design tools that based on only partially overlapping criteria (Henschel et al, 2004; Wang and Mu, 2004; Chalk et al, 2005). SiRNA vendors have also made their own siRNA design software available on the web. |
| One common drawback of all currently existing siRNA design software is that they are all based on limited data sets about siRNA efficacy. It is anticipated that, with the development of high throughput siRNA construction and validation methods, algorithms based on randomly chosen siRNA will emerge to provide better predictions for functional siRNA. |
| Although a siRNA library can be constructed by chemical synthesis, as done by Qiagen for Novartis and more recently by Dharmacon, most of the siRNA libraries covering more than a couple of thousand of genes are done by vector-based approaches. This is because, i) a vector-based siRNA library can be regenerated with minimal effort, ii) the constructions can be carried out in a standard molecular biology laboratory, and iii) a vector-based siRNA library can be much more affordable than a chemically synthesized library. |
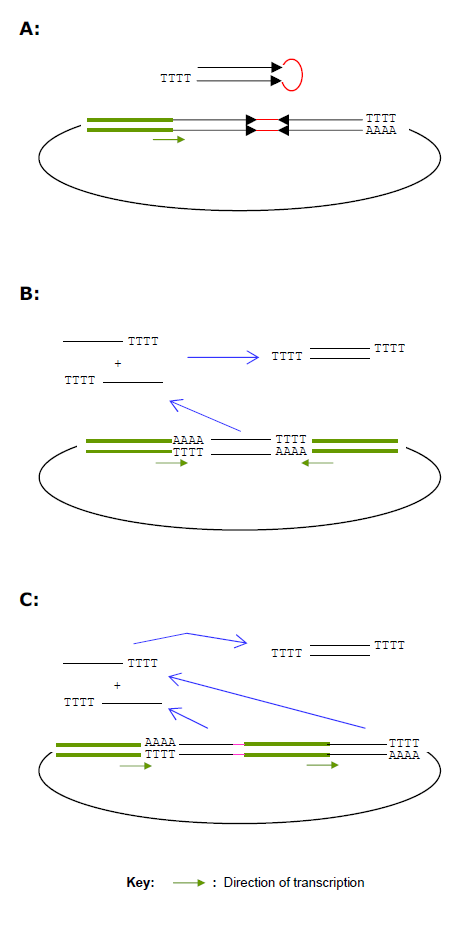 |
| Figure 1. Diagrams of three general ways of encoding siRNA in a plasmid or viral vector. The same principles apply also in PCR cassettes that derive from these vectors. A, a hairpin-like siRNA (shRNA) is generated by transcription from a segment of DNA (50-60 bp in length) driven by a single promoter for pol III RNA polymerases. B, expression of a siRNA from a single 19-mer DNA fragment through transcription driven two “opposing” promoters for Pol III RNA polymerases. C, expression of a siRNA from a two separate 19-mer DNA fragments through transcription driven two “tandem” promoters for Pol III RNA polymerases. |
| Three large-scale siRNA libraries have recently been constructed by academic researchers (Paddison et al, 2004; Berns et al 2004, Michiels et al, 2002) and Galapagos has generated the first large scale adenovirus based siRNA library in the industrial sector. Multiple efforts have been initiated since then by commercial companies, and notably among them, Genordia AB, a Sweden-based company will most likely be able to deliver the first siRNA library that covers a whole mammalian genome in 2005.` |
| Hannon and colleagues have constructed a large-scale siRNA library targeting a selected panel of human and mouse genes using a hairpin siRNA encoding cassette (Berns et al, 2004). The siRNA-encoding cassette was first cloned into a pSHAG-Magic donor vector. Although in most siRNA applications people has chosen to use 19- 21mer siRNA, this particular siRNA library encoded siRNA expression unit that contained 29 nucleotides, simple loop structure and U6 promoter. It was found that 29- nucleotide hairpins were more effective than shorter hairpin siRNA in this study. Since the efficacy of short siRNA is well established, this interesting finding might need more independent confirmation before being adopted as a guideline for siRNA libraries construction. Also concerns have been raised for possibly higher level of off-target hits as well as induction of antiviral responses. Totally 28,659 shRNAs targeting 9,610 human genes and 9,119 shRNAs targeting 5,563 mouse genes were included in the Hannon siRNA library. Some characteristics of this library include: 1) all clones were sequence verified. The extensive sequence verification was warranted because 25–75% of cloned shRNAs were found to contain significant mutations that arose during chemical synthesis. 2) Only coding sequences were targeted, and each shRNA was chosen such that it contained not more than 26 bp matches to any other gene. It should be cautioned that recent studies have demonstrated that for processed siRNA sequence identity up to 16-17 might already allow the siRNA to have offtarget effects. 3) Where possible, shRNAs were designed to have sequence identity to the mouse orthologue of the targeted gene. In terms of gene family representation, this library covers about 85% of all known kinases and phosphatases by three or more siRNAs per gene. Most other functional classes contain between 30–60% coverage with three or more hairpins, and 80% of genes by at least one sequence-verified hairpin. A unique 60-nuclotide DNA bar code has been integrated to each vector to allow identification of shRNAs in populations of virally transduced cells through hybridization methods. |
| The human RNAi library (the 'NKi library') constructed by Bernards and colleagues contains 23,742 siRNA vectors that cover 7,914 human genes (Michiels et al, 2002). Gene within this collection include components of major cellular pathways, such as the cell cycle, transcription regulation, stress signaling, signal transduction and important biological processes such as biosynthesis, proteolysis and metabolism. In addition, genes implicated in cancer and other diseases are included in the library. The siRNA encoding DNA fragments were cloned in a high-throughput fashion into pRetroSuper (pRS), a previously reported retroviral vector containing the shRNA expression cassette. This is one of the first large scale siRNA collection in the academic sector and in the validation of this library, researchers have achieved on average 70% expression inhibition for approximately 70% of the genes in the library using a pool of three knockdown vectors against a single gene. Compared to siRNA library made in plasmid vectors, siRNA librarys constructed in viral vectors has to be packaged and mass produced before being used in any knockdown experiments. These might comprise a major limitation in the application of viral vector based siRNA libraries in very large-scale screening. |
| Schultz and his colleagues have developed a PCR based system to generate siRNA for about 8000 genes at 2- siRNA/gene coverage using an opposing promoter approach. The siRNA has been used to discover novel members of the NF-kB pathway (Zheng et al, 2004). A singlestep PCR protocol was used to produce siRNA expression cassettes in a high-throughput fashion and showed that these siRNA expression cassettes can also specifically and efficiently suppress the expression of both transfected and endogenous genes. This single-step PCR approach is efficient and cost-effective and makes high-throughput production of siRNA expression cassette libraries for genome- wide functional gene annotation practical. The arrayed library was used to screen for genes involved in the NF-B signalling pathway that controls many diverse cellular processes including growth, development, inflammation, immune response, apoptosis, and oncogenesis. |
| Notably, one of the first viral based siRNA libraries was generated by an industrial player, Galapagos Genomics. This adenoviral siRNA library contains knock-down reagents for over 4,900 human druggable transcripts, including G-protein coupled receptors (GPCRs), ion channels, nuclear hormone receptors, kinases, phosphodiesterases and other druggable transcripts. Three adenoviral vector encoded siRNAs were designed for each gene. This threefold redundancy in siRNA sequences gives a 90% probability that the mRNA for any given gene will be knockeddown by at least 75%. Such a loss-of-function gene collection provides an ideal complement to the arrayed adenoviral gain-of-function libraries from the same source. Other characteristics of this siRNA library include: 1) the siRNA expression cassette is based on the human U6 promoter, a strong, ubiquitously active promoter that lacks essential promoter elements within the transcribed region. 2) The U6 promoter-based expression cassette was cloned in an adenoviral adapter plasmid at the position of the E1 region. The adenoviral vectors were based on adenovirus serotype 5 with deleted E1 and E2A regions. 3) The viral vectors were found to be able to express processed hairpin RNAs for at least 10 d. 4) The adenoviral vectors used have a broad tropism and very efficiently infect many different primary cell types including primary keratinocytes and primary synoviocytes, two cell types that are notoriously difficult to transfect. The use of siRNA tools in primary cells has been hampered by inefficient transfection protocols. Advantages in the transfection aspect are of critical importance for some applications, and thus counter-balance the burden imposed by the cumbersome viral handling. |
siRNA libraries with degenerated sequences |
| In a conventional siRNA application, a target gene is chosen first, and then siRNA sequences are designed against the transcript of this gene. In this approach, the choice of gene, and choice of targeting sites are inevitably biased due to the limited number of genes and sites that can be included in a particular study. The bias also arise inevitably from the fact that we have just started to get a relatively complete picture of the part of the mouse transcriptome that has a poly (A) tail, and knowing little about transcripts that do not have a such a tail. The development of siRNA expressing libraries that encode substantially all permutations of 19-mer siRNA thus become an appealing approach to allow the use of it as a single affordable tool for un-biased genome-wide screening. |
| Towards this direction, siRNA libraries have been synthesized by integration of 19-mer fully randomized DNA sequences into different siRNA encoding vectors (Chen et al, 2005). Such vectors should have theoretical complexity of 2.75x1011 in order to encode all the permutations of siRNA. This is a complexity that, albeit being reachable, will impose significant burden on the manipulation of the library and use of it for screening. Lucky or not, the siRNA was actually found to be tolerant to mismatches towards its terminus. This makes it reasonable to assume that a library will about 1 x 109 complexity will cover all possible siRNA. Such a complexity then comes in a range that is practical for plasmid library construction and maintenance. The library was used for phenotype-driven screening using cell proliferation as a model, and multiple siRNA that can induce significant enhancement of cell growth were identified. |
| Another alternative approach is to generate semi-random siRNA library from an mRNA source. In this case, a specific mRNA population is converted into double stranded cDNA, which will be cut into short DNA fragments that can then inserted into the vectors for encoding siRNA. The advantage of this type of libraries is that it will only have a complexity of in the neighborhood of 106 but already be able to cover a substantial part of the genome. The limiting aspect of the approach is that a library made in this way will only be useful for the same organism, or even only for the same cell type, whereas other fully randomized libraries can be used in all cell types and all relevant organisms. There have been several reports (Luo et al, 2004; Sen et al, 2004; Shirane et al, 2004) for the construction of such semi-random siRNA libraries and one of these is briefly described here as an example (Sen et al, 2004). In this method, mRNA is converted into double-stranded cDNA mixture that was further cleaved with a mixture of five restriction endonucleases, each of which leaves a 5'-CG overhang but has a different 4-bp recognition sequence. This combination covers 6 of 256 possible 4-bp sequences, resulting in one cleavage every 43 bp. These fragmented cDNAs are ligated to a hairpin linker with a complementary 5'-CG overhang. This hairpin has a recognition site for MmeI, a type II restriction endonuclease that cleaves 20–21 bp away from its recognition sequence and leaves a two-base 5' overhang. A hairpin linker is then ligated to either the 5' or 3' end of the double-stranded cDNA fragment. Next an "extension linker" comprised of three different oligonucleotides is ligated to the cDNA-extended hairpin. As a result the 5' and 3' ends of the extended hairpin become ligated to the extension "sense" oligonucleo tide (green) and the extension "antisense" oligonucleotide respectively. The extension priming oligonucleotide serves as primer for DNA elongation by the highly processive Bst DNA polymerase large fragment, which lacks a 5'–3' exonuclease domain. This reaction converts the hairpin into a dsDNA that encodes a hairpin RNA. The process has been used to construct a siRNA library from a mouse embryo cDNA library. This resulted in three million independent clones. Twenty-seven clones were chosen at random for sequencing, all of which revealed matches against known mouse genes. Five random clones were used to generate stable NIH 3T3 cell lines. Northern blotting with probes corresponding to their mRNA targets demonstrated that each of the cDNA-derived inserts produced the expected processed 21-base siRNAs and these clones caused specific reductions in their corresponding target mRNA. |
| Functional screening using different siRNA libraries |
| Genome-wide RNAi screens were explored in C. elegans using libraries of in vitro-transcribed long dsRNAs or simply the bacteria that express dsRNA. For example, an RNAi feeding library consisting of 16,757 bacterial clones covering majority of the worm genome was constructed (Fraser et al, 2000; Kamath et al, 2003). Upon feeding the worms, these clones generated transient loss-of-function phenotypes for many genes by inactivating the target genes via RNAi. Genome-wide RNAi approaches have been used successfully for phenotype-based screens also in Drosophila melanogaster. In part, these successes derived from the availability of convenient and inexpensive methods for producing and introducing dsRNA. To date, the use of RNAi libraries in mammalian systems is becoming well accepted but most of the work has been limited to specific protein families because of technical and practical issues associated with generating large synthetic siRNA and/or vector-based short hairpin RNA (shRNA) expression libraries, but the situation is rapidly improving with the technology development mentioned above. Screening schemes using large-scale siRNA libraries are understandably diverse due to differences in aims, vector systems, and read-out methods. The five case studies presented below could give us a hint about how such libraries can be deployed in HTS. |
| Retroviral vectors can be used to transfect a large variety of cell types, and its short-coming on the handling side can be well balanced by using it in a combinatorial manner, e.g., in libraries. Work done by Berns et al (2004) has provided a typical example of such applications in a medium scale. In this study, retroviral vectors were generated in pools and then the pools were used to transfect BJ-TERTtsLT cells at 32 °C. After two days’ incubation at 32 °C, the incubation temperature was increased to 39 °C for proliferating colony screening. In the first-round screening, six pools of infected cells were found able to form colonies at 39°C. These shRNA inserts were then re-cloned from these colonies, and their identity was established by DNA sequence analysis. Only those shRNA inserts that were presented in multiple independently derived colonies were further analyzed in the second-round screening in BJT ERT-tsLT fibroblast. Using this approach, shRNAs against six genes were identified that could suppress the temperature-shift-induced proliferation arrest in the BJ BJT ERT-tsLT cells. Remarkably one identified gene was p53, which underscores the quality of the NKi library. Knockdown of other five identified genes also mediated escape from proliferation arrest in BJ-TERT-tsLT cells when tested in the absence of retroviral vector, suggesting that inhibition of these genes allowed bypass of the p53 response. In screening as such, it would always be a concern whether the change in read-out is a consequence of on-target hit or off-target hits. In this case, it was found that two of the three shRNAs targeting a single gene could mediate escape from growth arrest for three of the five newly identified genes. This could be considered as a confirmation that the effects of the siRNAs were 'on-target'. For genes that only have a single siRNA hit, normally additional siRNA need to be designed to confirm any phenotypical changes observed with the initial siRNA clone. |
| In a similar screening, 6712 shRNA expression targeting 4873 genes were used to discover novel members of proteasome. The 26S proteasome is the major non-lysosomal protease in eukaryotic cells. To search the library for shRNAs that compromise proteasome function, a reporter assay was used in which a fluorescent protein (Zoanthus green fluorescent, ZsGreen) was coupled to a well characterized degradation signal, the mouse ornithine decarboxylase (MODC) gene. Another plasmid encoded Discosoma red fluorescent protein (DsRed) was used as a normalization control for transfection. Individual transfection of 6,712 shRNAs revealed approximately 100 RNAi constructs that increased the accumulation of ZsGreen– MODC, of which 22 corresponded to 15 known proteasome subunits. |
| As mentioned above, a siRNA expression cassette library targeting 8,000 human genes from the public UniGene library were generated in Schultz lab in Scripps Institute in a vector containing dual pol III promoters, with two targeting sequences per gene Zheng et al, 2004). To screen for regulators of NF-kB transcriptional activation by TNFa, the pNF-B-Luc reporter plasmid was cotransfected with individual siRNA expression cassettes into HEK293T cells. Of 94 genes identified in the initial screening, 20 hits were further assessed by introducing additional siRNAs, and 17 of them seemed to be genuine on-target hits. Among them, eight are known to be involved in the NFkB signalling. Of the remaining genes, some might be involved in cellular processes that indirectly affect the NF-B pathway, whereas others might have previously unrecognized roles in the NF-B signalling pathway. |
| All screening work mentioned above were done in microtitre plates, which will remain a major screening platform in the future. Cell microarrays were created to provide much higher throughput than microtitre plates for siRNA based screening. Cell microarray was first described by Ziauddin and Sabatini (2001) who demonstrated that cells grown on a glass substrate could take up DNA–lipid complexes that had been deposited on the slide before cells were plated. Cells became transfected in situ, with defined spots of transfected cells localized over the printed DNAs. Cell microarrays represent a novel alternative to classical approaches to phenotype-based assays in mammalian cells, at least for some of the easy-to-transfect cells such as HEK 293T, IMR90/E1A, NIH 3T3, and HeLa cells. In this screening format, siRNA (plasmids or PCR cassettes) are spotted onto glass slides together with transfection reagents (normally with gelatin too for immobilization purpose). The vector based siRNA reagents appear to be stable for months on such chips. When cells are overlaid on top of a spot on the slides, corresponding siRNA vector will be taken into the cells so that siRNA will be expressed, and the effect of the siRNA can be read out in high throughput according to the phenotypes or the reporter systems used. The initial proof-of principle was done by siRNAs corresponding to the enhanced Green Fluorescent Protein (GFP) gene (eGFP) on microscope slides, and plated HeLa cells permanently expressing a destabilized version of eGFP on top of the slides. The result showed the cellular uptake, "reverse transfection", of rhodamine-tagged siRNA (rh-eGFP), and silencing of GFP expression in cells. An experimental comparison was provided by carrying out, side by side, the array-based and microtitre plate screening of siRNA that can perturb proteosome functions. The result showed that the array method does work in screening for siRNA that can manipulate endogenous genes, and likely to provide data that is even better than the classic microtitre plate based screening process. Advantage of the method is that large number of siRNA can be rapidly screening through a given phenotype or reporter system. The limitation of the method is, in additional to the need of complex instrumentation, that many phenotypes can’t be fit into such an array- based read-out format. |
Conclusions |
| RNAi provides a perfect example of how a natural occurring process can be “hijacked” by researchers and used as a research tool. The penetration speed of this technology into the labs in both academic and industrial settings has been astonishing even though the mechanism of RNAi has not been fully understood. In this reveal, we tried to focus on the development and application of vector based siRNA libraries due to that fact that although synthetic siRNA has become much more affordable now than two years ago due to stiff competition and improvement in the throughput of RNA synthesis, vector-based siRNA will still be the reagent of choice for large scale siRNA based screening in most academic institutions and companies. It is worth to stress that synthetic siRNA has also certain advantages over vector-based siRNA approaches in dosing, integration of modified nucleotides, and delivery, in just name a few aspects for example. |
| It is anticipated that plasmid libraries that have a genome- wide coverage will become available in 2005 for screening using both transient transfection as well as stable transfections. Inducible promoters will be integrated into some of the libraries for special screening need. While lentivirus and other viral vectors provide obvious advantages in the transfection of hard-totransfect cells, they are bound to be significantly cumbersome to work with in a high throughput screening. It is more likely that they will serve as second line of verification tools instead of being used as the first line screening tools in many occasions other than combinatorial screening. |
Acknowledgements |
| The authors wish to acknowledge Pfizer Inc and the Chinese High Tech Research and Development Program (863) for support. |
Statement Of Competing Interests |
| Authors Hong-Yan Zhang, Claes Wahlestedt and Zicai Liang are founders of a reagent company, Genordia AB, specializing in the development, production and sales of RNAi-related products. |
References
|