- Journal of RNA and Genomics (2006) Review Article
Manipulating and enhancing the RNAi response
| Peter I Joyce*, Joseph M Gallagher and Patricia E Kuwabara Department of Biochemistry, School of Medical Sciences, University of Bristol, Bristol, BS8 1TD, UK |
| Correspondence to:Patricia E Kuwabara, Email: P.Kuwabara@bristol.ac.uk, Tel: +44 117 3317275, Fax: +44 117 9288274 |
| (Received 20 December 2005; Accepted 03 January 2006, Available online 12 January 2006; Published 28 February 2006) |
Abstract
The phenomenon that is known as RNA mediated interference (RNAi) was first observed in the nematode C. elegans. The application of RNAi has now been widely disseminated and the mechanisms underlying the pathway have been uncovered using both genetics and biochemistry. In the worm, it has been demonstrated that RNAi is easily adapted to high throughput analysis and screening protocols. Hence, given the availability of whole genome sequences, RNAi has been used extensively as a tool for annotating gene function. Genetic screens performed with C. elegans have also led to the identification of genes that are essential for RNAi or that modulate the RNAi process. The identification of such genes has made it possible to manipulate and enhance the RNAi response. Moreover, many of the genes identified in C. elegans have been conserved in other organisms. Thus, opportunities are available for researchers to take advantage of the insights gained from the worm and apply them to their own systems in order to improve the efficiency and potency of the RNAi response.
Keywords |
| C. elegans, RdRP, RNA interference, siRNA, systemic RNAi |
Introduction |
| RNA mediated interference (RNAi) is extensively used as a sequence-specific tool for generating knock-down phenotypes and examining gene function in a wide variety of organisms. It also displays promise as a technique for therapeutic intervention in human disease. In the nematode C. elegans, investigations into the mechanisms underlying RNAi were instigated by the paradoxical finding that the germline injection of in vitro synthesised RNA corresponding to either the sense or antisense strand of par-1 mRNA mimicked the par-1 loss of function phenotype (Guo and Kemphues, 1995). The response elicited by exogenous sense strand par-1 RNA indicated that the inhibition of cognate par-1 activity could not be caused simply by translational inhibition promoted by base pairing between the injected and endogenous par-1 mRNA. The resolution of this paradox was provided by Fire and colleagues; they demonstrated that double stranded RNA (dsRNA) was a substantially more potent agent for inhibiting gene activity than either single-stranded sense or antisense RNA (Fire et al, 1998). It is now known that the RNAi pathway of C. elegans shares mechanistic similarities with post transcriptional gene silencing (PTGS) in plants and fungi (Cogoni and Macino, 1999; Baulcombe, 2004) and homology-dependent gene silencing in higher organisms (Hammond et al, 2000; Wianny and Zernicka- Goetz, 2000). |
| This review will focus on using RNAi as a tool for analysing gene function, in which the RNAi response is triggered by the addition of sequence-specific exogenous dsRNA or short interfering RNA (siRNA). In particular, we will describe how the genetic tractability of C. elegans has been used to great advantage not only to identify genes that are essential for RNAi, but also to identify genes that modulate or negatively regulate the pathway. Because many of the key components of the RNAi pathway are conserved across phyla, insights gained from the worm have the potential to be translated to other organisms and enhance both the efficacy and potency of RNAi as a tool for studying gene function (Hamilton and Baulcombe, 1999; Caplen et al, 2001; Elbashir et al, 2001; Knight and Bass, 2002; Kennedy et al, 2004; Wang et al, 2005). |
The RNAI Pathway in C. ELEGANS |
| Obviously RNAi is not simply a tool intelligently designed for researchers to knockdown gene activity. Moreover, the number of processes involving components of the RNAi machinery has burgeoned; RNAi is involved in a variety of basic physiological responses ranging from the control of gene expression to the establishment of heterochromatin silencing. In C. elegans, it has been suggested that the natural physiological function of RNAi is to protect C. elegans against viral infection or genomic invasion by transposable elements (Lu et al, 2005; Wilkins et al, 2005). It has also been shown that C. elegans transgenes, which often form large tandem arrays, can lead to co-suppression of both the transgene and its chromosomal homologue through RNAi (Ketting et al, 1999; Dernburg et al, 2000). Components of the RNAi machinery also participate in the processing of microRNAs (miRNAs),which are considered to be natural substrates for Dicer (Zamore and Haley, 2005). It has been shown that miRNAs can target mRNAs for degradation or inhibit the translation of mRNAs by binding to sites in the 3’ UTR (Ambros, 2001; Bagga et al, 2005). In yeast and Drosophila, RNAi has been shown to play a role in establishing domains of heterochromatin (Hall et al, 2002; Volpe et al, 2002; Pal-Bhadra et al, 2004; Verdel et al, 2004). Here, however, we will limit our discussion to the classical pathway of RNAi in C. elegans, which is triggered by the addition of exogenous dsRNA. |
Initiation of RNAi BY dsRNA |
| The basic mechanics underlying RNAi have been uncovered using a combination of genetic and biochemical studies in C. elegans and Drosophila. The presence of short interfering RNAs (siRNAs) was first observed in plants (Hamilton and Baulcombe, 1999). Subsequently, siRNAs have been found in Drosophila S2 cell extracts and in C. elegans (Hammond et al, 2000; Zamore et al, 2000). In mammals, RNAi is initiated by the addition of siRNAs, because introducing large dsRNA fragments activate an interferon driven inhibition of translation (Samuel, 2001). By contrast, both the worm and Drosophila lack an interferon response, which makes it is possible to trigger RNAi by introducing relatively large dsRNA fragments (500-1000 bp), possibly corresponding to the entire length of an mRNA. |
| How are siRNAs generated from large dsRNA molecules in the worm? Screens carried out by Tabara and colleagues led to the discovery of the first RNAi deficient (rde) pathway mutants in C. elegans (Tabara et al, 1999). Two of these genes, rde-1 and rde-4, encode interacting proteins; rde-4 encodes a dsRNA binding protein and rde-1 encodes a PAZ-PIWI/Argonaute protein. RDE-4 appears to promote the specific recognition of foreign dsRNA, because it does not interact with mRNA or dsRNA derived from an amplification process, which will be discussed below (Parrish and Fire, 2001). RDE-4 also interacts with the conserved DExH-box helicase, DRH-1 (Figure 1) (Tabara et al, 2002). Together these proteins form a complex with dicer (DCR-1), a dsRNA specific RNaseIII ribonuclease, which is responsible for cleaving dsRNA into 21-25 nt siRNAs (Tabara et al, 2002; Meister and Tuschl, 2004). |
Risc Nuclease Activity |
| Early studies in C. elegans using in situ hybridisation revealed that dsRNA mediated interference led to a marked reduction in target mRNA transcripts (Montgomery et al, 1998). This observation supported the existence of a sequence- specific nuclease complex, which was responsible for cleaving and degrading target mRNA before it was translated (Montgomery et al, 1998). |
| Physical evidence supporting the existence of this complex was first obtained using a Drosophila cell-free model system to identify a preformed entity with nuclease activity, named RISC (for RNAi induced silencing complex). In Drosophila, RISC nuclease activity consists of the proteins Tudor-SN, VIG, Argonaute 2 and FXR bound together with siRNAs (Hammond et al, 2000; Caudy et al, 2002; Caudy et al, 2003). C. elegans orthologues of Tudor-SN and VIG, named TSN-1 and VIG-1, respectively, have been identified and shown to share similar functions as Drosophila and mammalian proteins (Figure 1) (Caudy et al, 2003). TSN-1contains five staphylococcal/micrococcal nuclease domain repeats, the last of which is fused to a tudor domain. Although TSN-1 appeared to be an obvious candidate for Slicer, the nuclease that cleaves siRNA targeted mRNA, this now appears unlikely. However, TSN-1 could still play a role in degrading target mRNA; TSN-1 is also likely to have activities that are distinct from its participation in RISC, because it has both a nuclear and cytosolic location and can cleave both RNA and DNA (Caudy et al, 2003). VIG-1 carries an RGG box, which is a motif involved in RNA binding; however little is known about its function in RISC (Caudy et al, 2002). |
| In Drosophila and mammals, RISC contains a PAZ-PIWI Argonaute protein, Ago-2. The crystal structure of the PAZ domain of Ago-2 shows the presence of an RNA binding domain, which is important for siRNA interactions (Song et al, 2003). Additional structural studies revealed that the PIWI domain contains an RNaseH fold, which makes it capable of performing endonucleolytic cleavage of mRNA targeted by siRNAs (Parker et al, 2004; Song et al, 2004). It was further confirmed that Ago-2 is Slicer through reconstitution studies (Rivas et al, 2005). In C. elegans, an Argonaute protein associated with RISC has not been identified, although RDE-1 is a PAZ-PIWI/ Argonaute protein involved in the initiation of RNAi. However, there are 25 predicted PAZ-PIWI homologues in C. elegans, which might act redundantly in RISC (Tabara et al, 1999); one of these PAZ-PIWI homologues, PPW-1, is required for germ line RNAi (Figure 1) (Tijsterman et al, 2002b). |
Amplification and the Production of Secondary siRNA |
| In C. elegans, RNAi acts catalytically - only a few molecules of dsRNA are required to trigger the silencing of a much larger amount of mRNA (Fire et al, 1998). Although, cleavage of the dsRNA trigger by Dicer into siRNAs provides some amplification, it does not fully account for the stoichiometry of the amplification. In further support of an amplification step in C. elegans RNAi, secondary siRNAs that do not correspond to the input dsRNA trigger have been discovered. These secondary siRNAs, although different to the input trigger dsRNA, are identical with 5’ segments of the target mRNA (Sijen et al, 2001). It has been shown that siRNAs produced by the dicing of exogenous dsRNA can subsequently be used as primers to amplify a dsRNA from mRNA targets; the dsRNA thereby synthesized can then be presented to Dicer in order to produce a secondary set of siRNAs, which can cycle back to RISC to perform transitive RNAi (Figure 1). |
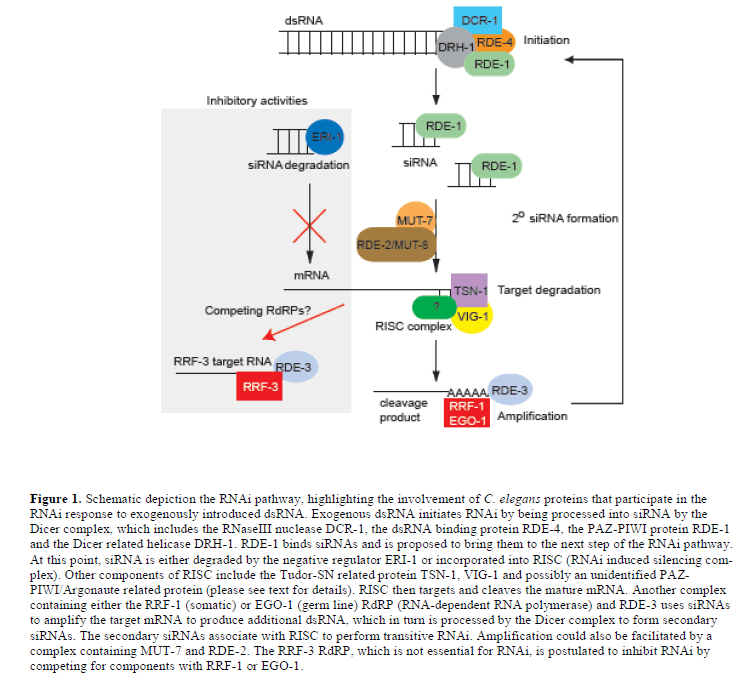 |
| It has been found that two of the four RNA-directed RNA polymerases (RdRPs) in C. elegans are essential for the RNAi amplification process. In the soma, the RdRP family gene, rrf-1 is required for production of secondary siRNA, which involves the priming of the mature target mRNA by the primary siRNAs (Sijen et al, 2001). In the germ line of C. elegans, the RdRP, EGO-1, plays role similar to RRF-, but also has essential roles in Notch signalling and oogenesis (Smardon et al, 2000). |
| The C. elegans MUT-7 protein, which is a putative exoribonuclease, and its interaction partner RDE-2 are also required in vivo for efficient RNAi (Tabara et al, 1999); these proteins form a complex that increases in size in response to RNAi activation (Tops et al, 2005). Both mut-7 and rde-2 are not only defective in RNAi, but they are also defective in transposon silencing as well as co-suppression (Tijsterman et al, 2002a; Sijen and Plasterk, 2003). When either mut-7 or rde-2 is absent, siRNAs fail to accumulate in vivo. In addition, mut-7 and rde-2 function downstream of rde-1 and rde-4. Taken together, these observations indicate mut-7 and rde-2 function downstream of Dicer and upstream of RISC, and are possibly involved in siRNA amplification. |
| Another mutant that fails to accumulate siRNAs and is also proposed to function in siRNA amplification is rde-3. RDE-3 is member of the polymerase ß nucleotidyltransferase superfamily. It has been proposed that RDE-3 might function by polyadenylating and stabilising the 5’ mRNA cleavage product generated when RISC is associated with primary siRNAs; in turn, the stabilised RNA could act as a template for an RdRP to generate secondary siRNAs (Figure 1) (Chen et al, 2005). |
rrf-3, The First Endogenous Inhibitor of RNAi |
| It is clear that RNAi is a powerful tool for analysing gene function, but as with any technique, there is always room for improvement or modification. Previous studies and anecdotal reports have shown that the nervous system of C. elegans is partially resistant to RNAi (Timmons et al, 2001). High throughput RNAi screens performed using wild type C. elegans (N2 Bristol) have also highlighted areas in which the efficacy of RNAi is sometimes limited. For example, 10% of the genes analysed displayed loss-offunction phenotypes; however, many genes that had previously been shown to have essential functions, particularly those that function in the nervous system, appeared to be less susceptible to dsRNA inhibition when administered by feeding or soaking (Fraser et al, 2000; Gonczy et al, 2000; Maeda et al, 2001; Timmons et al, 2001; Piano et al, 2002). In addition, it has been revealed that the consistency and reproducibility of high throughput RNAi screens could be improved by making weak or marginal RNAi responses more robust. |
| In the course of analysing activities of the four RdRP encoding genes in C. elegans, it was observed that an rrf-3 deletion mutant displayed increased sensitivity to RNAi (Sijen et al, 2001). Large-scale screens using rrf-3 mutant animals confirmed that the absence of rrf-3 activity led to an obvious enhancement in the sensitivity of a range of target genes, including neuronal genes, to RNAi (Simmer et al, 2002). One hypothesis is that RRF-3 normally has an inhibitory effect on RNAi because it competes with the somatic RRF-1 or germline EGO-1 RdRPs for dsRNA substrates (Figure 1) (Simmer et al, 2002). Despite the essential role for the EGO-1 RdRP in the germ line of C. elegans, genes encoding RdRP orthologues have not been found in either the Drosophila or mammalian genomes, although an RdRP activity has been identified in mouse erthyroleukaemia cells (Volloch et al, 1987; Stein et al, 2003). It is possible that another RNA polymerase could perform the activity of an absent RdRP, however, it is also possible that mammalian cells do not require an amplification step for effective RNAi. In C. elegans, the absence of rrf-1 mediated amplification can be partially suppressed by knocking out eri-1, an RNAi antagonist that is described in the next section (Kennedy et al, 2004). |
eri-1, A Conserved Antagonist of RNAi |
| The systemic nature of RNAi in C. elegans and plants - the ability of the RNAi effect to be propagated across cell boundaries - has been exploited to great advantage. Not only can RNAi be performed by injecting dsRNA almost anywhere in the animal, but it is also possible simply to feed worms Escherichia coli expressing gene-specific dsRNA (Timmons and Fire, 1998) or to soak them in solutions of dsRNA (Tabara et al, 1998). The ease by which RNAi can be performed in C. elegans has made it feasible to undertake high throughput whole genome screens designed to uncover gene function and aid in genome annotation (Fraser et al, 2000; Gonczy et al, 2000; Maeda et al, 2001; Timmons et al, 2001; Piano et al, 2002) |
| The ability to identify gene mutations capable of either inhibiting or enhancing the effectiveness of RNAi led researchers to perform targeted mutant screens aimed at identifying additional genes that could enhance the effectiveness of RNAi. One such screen, which was designed to identify mutants with enhanced sensitivity to RNAi in the nervous system of C. elegans, identified eri-1 (Kennedy et al, 2004). The absence of eri-1 activity increased the effectiveness of RNAi in most tissues, not only the nervous system, although eri-1 is preferentially expressed in neurons and the somatic gonad. The ERI-1 protein is a member of the DEMDh exonuclease subfamily, which belongs to the DEDDh family of exonucleases. ERI-1 contains a SAP domain found in DNA binding proteins and a DEDDh-like 3’ J 5’ exonuclease domain, which is also found in RNases such as RNase T and oligoribonuclease (Kennedy et al, 2004). In vitro studies show that this domain can degrade the 3’ overhangs of siRNA molecules and prevent their incorporation into RISC (Figure 1); thus, ERI-1 could normally act as an RNAi inhibitor that reduces the degradation of target mRNA (Kennedy et al, 2004). |
| eri-1 orthologues have been identified in higher organisms and also appear to act as inhibitors of the RNAi pathway (Table 1). It has been demonstrated that removal of meri-1, the mouse orthologue of eri-1, by siRNAi can enhance the sensitivity of RNAi (Hong et al, 2005). Mice that carry the reporter plasmid pCMV-iHBS produce hepatitis B virus serum antigen (HBsAg). However, when these mice are injected with siRNAs corresponding to meri-1 and HBVP, a significant decrease in both the secretion of HBsAg and in the level of meri-1 transcript are observed, when compared to a similar cohort treated only with siRNAs to target HBVP. So, as in C. elegans, it appears that inhibition of meri-1 enhances RNAi. Moreover, when mice were exposed to high doses of siRNA, meri-1 expression was up-regulated, suggesting increased degradation of siRNA with greater concentrations of siRNA. |
| In humans, the eri-1 orthologue, 3’hExo, is involved in processing histone mRNAs with stem-loop structures, and is a major regulator of histone mRNA biogenesis and metabolism (Dominski et al, 2003). It has also been demonstrated that siRNAs are also a substrate for 3’hExo (Kennedy et al, 2004). When HeLa cells are co-transfected with both a minigene that expresses the T cell receptor-ß (TCR-β) and a transgene that expresses a short hairpin RNA (shRNA) that targets the RNAi-mediated degradation of TCR-ß mRNA, overexpression of 3’hExo increases the degradation of TCR-ß mRNA by 8-fold (Buhler et al, 2005). Taken together, these results indicate ERI-1, MERI-1 and 3’hExo share a conserved function as an siRNase. The identification of eri-1 in C. elegans and its conservation as a siRNase in RNAi, suggests that elimination of eri-1 in mammalian cells could increase the efficacy of RNAi in these systems. |
Systemic RNAI in C. ELEGANS |
| The systemic nature of C. elegans RNAi was already fully realised in the original report for Fire et al. (1998). To identify genes that facilitate systemic RNAi, directed screens were performed by two groups and sid-1 (systemic RNAi defective)/rsd-8 (RNAi spreading defective) was obtained (Winston et al, 2002; Tijsterman et al, 2004). sid- 1 encodes a 776 amino acid protein, containing 11 transmembrane domains and a large extracellular N-terminal domain (Winston et al, 2002; Tijsterman et al, 2004). sid-1 localises to the cell periphery in cells exposed to the environment, including some but not all neurons, and is essential for cell-autonomous RNAi in C. elegans (Winston et al, 2002; Tijsterman et al, 2004). Transfection of SID-1 into Drosophila S2 cells reveals that SID-1 promotes the import of dsRNA in a process that is based on passive diffusion (Feinberg and Hunter, 2003). It has also been proposed that sid-1 could indirectly promote the uptake of dsRNA by modifying the properties of some other transporter. The molecule being transported appears to be dsRNA and not siRNA (Tijsterman et al, 2002a). The efficiency of this transport system appears to be dependent on the length of dsRNA; longer dsRNAs are transported with greater efficiency than 21 bp siRNAs (Feinberg and Hunter, 2003). |
| Orthologues of SID-1 have been found in insects and mammals. It has recently been demonstrated that systemic RNAi gene silencing also exists in the grasshopper, Schistocerca americana. Injection into the dorsal heart vessel of first instar nymphs of dsRNA to the vermillon (Sa_v) gene, which confers eye colour, resulted in red eye colouration consistent with an RNAi knockdown (Dong and Friedrich, 2005). The presence of a ubiquitously expressed sid-1 homologue (Sa_sid-1) suggests that sid-1 might play a conserved role in mediating systemic RNAi in animals. |
| The mammalian sid-1 homologue, SIDT1, which similarly encodes a protein with 11 predicted transmembrane domains, also localises at the cell periphery (Duxbury et al, 2005). By contrast to Ce-SID-1, which does not transport siRNAs very efficiently, overexpression of SIDT1 increases the uptake of siRNA via soaking, a process that also appears to occur through passive diffusion, and causes increased siRNA mediated knockdown (Duxbury et al, 2005). Thus, it is possible to consider performing large scale RNAi, perhaps by taking advantage of cell based microarrays and using cell lines that have enhanced RNAi sensitivity due to overexpression of the mammalian SIDT1 (Wheeler et al, 2005). |
| Mutations in rsd-3 that render mutants defective in systemic RNAi have also been identified (Tijsterman et al, 2004). The rsd-3 gene encodes a protein with an epsin Nterminal homology (ENTH) domain; ENTH domains bind phosphoinositides, which are often present in vesicle trafficking domains (De Camilli et al, 2002). RSD-3 also shares homology with the human protein Enthoprotin (also known as Clint and Epsin-R) (Tijsterman et al, 2004). Enthoprotin co-localises with clathrin and the AP-1 adaptor protein and promotes clathrin dependent membrane budding from the trans-Golgi network (TGN) (Wasiak et al, 2002; Chidambaram et al, 2004). This raises the possibility that vesicle trafficking involving RSD-3 plays a role in systemic RNAi, possibly by mediating the transport and packaging of dsRNA and/or siRNA inside the cell. The connection between the roles of RSD-3 and SID-1 in systemic RNAi is presently unknown. However, it is likely that additional insights into the process of systemic RNAi will be provided by characterisation of other genes, which when mutated prevent systemic RNAi, such as rsd-2, rsd-3 and rsd-6 (Tijsterman et al, 2004). |
The Role of Adars in RNAi |
| Adenosine deaminases that act on RNA (ADARs) edit dsRNA by converting adenosines to inosines through deamination (Bass, 2002). Thus, RNA editing can produce multiple isoforms of the same primary transcript; the greater the extent of base-pairing, the more extensive the editing. One obvious implication of this process is that dsRNAs modified by ADARs could potentially escape recognition by Dicer; hence, ADARs could act to suppress RNAi. There are two ADARs in C. elegans, adr-1 and adr-2; however, contrary to initial expectations, the absence of adr activity failed to enhance the sensitivity of RNAi triggered by exogenous dsRNA. However, the absence of adr activity specifically enhanced RNAidependent co-suppression. It was found that when a transgene carrying a heatshock-driven short sequence-specific hairpin RNA (shRNA) was introduced into the adr-1;adr- 2 double mutant, heat-shock independent co-suppression was detected. Subsequently, it was shown that all transgenes introduced into adr-1;adr-2 null animals were silenced in somatic tissues. These observations led to the hypothesis that in the absence of RNA editing mediated by adr-1 and adr-2, leaky transcription from transgenes form dsRNA and initiate RNAi via Dicer (Knight and Bass, 2002). Therefore, ADR-1 and ADR-2 could play a protective role in preventing co-suppression in C. elegans; however, it appears that co-suppression can still occur if the system becomes overloaded with dsRNA from transgenes. It is probable that ADARs exert their activity on dsRNA generated from transgenes, but not on exogenous dsRNA because these proteins are primarily localised to the nucleus and not the cytoplasm (Billy et al, 2001). |
| Interestingly, animals lacking either adr-1 or adr-2 also suffer from olfactory defects in chemotaxis. Inhibiting RNAi by removing rde-1 or rde-4 alleviates this defect, suggesting that ADARs could play a special role in the nervous system by preventing endogenously expressed dsRNAs from activating RNAi (Tonkin and Bass, 2003). |
| Mammals have three ADARs (ADAR1 to ADAR3); ADAR1 forms two isoforms, ADAR1p150 and ADAR1p110 (for review, see Valente and Nishikura, 2005). Only ADAR1p150 displays a primarily cytoplasmic localisation (Patterson and Samuel, 1995). It has recently been found that ADAR1p150 binds siRNA containing 3’ overhangs with an extremely high affinity, but does not edit them (Yang et al, 2005); A-to-I editing by ADARs requires dsRNA of 30 bp or longer. Instead, it appears that ADAR1p150 acts to sequester siRNAs and prevent them from entering the RNAi pathway. In support of this model,Yang et al (2005) showed that the level of siRNA mediated gene silencing was significantly increased in ADAR1- /- mouse embryonic fibroblasts. In addition, ADAR1p150 is the only interferon inducible ADAR, which further suggests that ADARs could play a role in the natural RNAi response in mammalian cells (Patterson and Samuel, 1995). Expression of ADARs (and meri-1) is also upregulated in response to high concentrations of siRNA in mouse liver (Hong et al, 2005). In Xenopus oocytes, ADARs have been further implicated in RNAi because hyper-edited dsRNAs are associated with and degraded by the Tudor staphylococcal nuclease (TSN), which is a component of the RISC complex (Scadden, 2005). |
| Hence, studies in worms and vertebrates indicate that ADARs have definite, but distinct roles in RNAi. Unlike adr-1 and adr-2 in C. elegans, which normally inhibit RNAi mediated co-suppression caused by nuclear transgenes, the mammalian ADAR1p150 appears to play an inhibitory role in cytoplasmic RNAi. This raises the possibility that elimination of ADARp150 can be exploited as a method to improve the efficiency of performing RNAi screens in mammalian systems. |
Chromatin Remodelling And RNAi |
| The C. elegans lin-15B gene is a class B synthetic multivulva (synMuvB) gene. Class B genes encode the Retinoblastoma (Rb) tumour suppressor orthologue and other components of the Rb complex. Mutations in these genes were initially identified because animals carrying mutations in both synMuvA and synMuvB genes develop cell lineage transformations that cause a multivulva phenotype, whereas animals carrying a single mutation do not show this tranformation (Huang et al, 1994). |
| Recent studies reveal that C. elegans lacking Rb or Rb complex proteins encoded by lin-15B, lin-35, dpl-1, lin- 53, lin-9, lin-13 and hpl-2 show enhanced RNAi responses revealed by a decrease in the level of target mRNA, particularly in the nervous system (Wang et al, 2005). In addition, these animals show inappropriate expression of germline activities in somatic tissue, such as pgl-1, which encodes an RNA-binding component of germline-specific P-granules, and ectopic P-granule-like structures. How does inhibition of the Rb pathway enhance RNAi? One hypothesis provided by the authors is that de-repression of some RNAi components, which are normally restricted in their activities to the germ line, allows them to function and enhance RNAi in somatic tissues. Support for this model comes from the observation that co-suppression associated with a nuclear transgene, which is normally limited to the germ line, is now extended to somatic tissues. In addition, the absolute requirement for rrf-1 is relaxed in Rb pathway mutants, suggesting that the germline RdRP, EGO-1, can now act redundantly in somatic tissues. The de-repression of germline activities in somatic tissues also requires the inappropriate expression of a chromatin remodelling complex defined by MES-4 (Wang et al, 2005). |
| It has also been suggested that mutations in the Rb pathway could enhance RNAi by alleviating a competition between the RNAi silencing machinery and the chromatin silencing pathway, so that shared components are redirected to perform RNAi. In support of this idea, a mutation in the C. elegans hpl-2 gene, which encodes an orthologue of the methylated histone binding protein HP1, enhances RNAi and causes transgene de-silencing (Couteau et al, 2002; Wang et al, 2005). Moreover, hpl-2 has been shown to function as a synMuvB gene. In hpl-2 mutants, pgl-1 is also misexpressed, suggesting that it is the combined effect of inappropriately expressing germline components in the soma, as well as releasing RNAi components from chromatin remodelling complexes, which is responsible for the enhancement of RNAi. |
| Combining Rb pathway mutants with previously characterised RNAi enhancer mutants, such as eri-1 and rrf-3, synergistically improves RNAi, indicating that the pathways affected are independent. The potential for Rb pathway mutants to enhance RNAi in mammalian systems has yet to be analysed. However, the recent creation of an Rb knockout mouse makes it possible to examine RNAi enhancing effects within this system (Zhang et al, 2004). |
| In addition to the genes mentioned in this review, genetic screens have identified many more proteins with possible roles in RNAi, including Piwi/PAZ domain proteins, DEAH helicases, RNA binding/processing factors, chromatin associated factors, DNA recombination factors and nuclear import/export factors(Kim et al, 2005). The range of potential genes that impinge on the RNAi process demonstrates the complexity of RNAi and its many cell biological roles, which will remain a subject for future study. |
Conclusions |
| Since 1998, great strides have been made in elucidating the mechanisms underlying RNAi and in identifying the genes that participate in the execution and control of this process. Model organisms, such as C. elegans, have played vital roles in helping to identify and deduce the function of genes involved in RNAi and to order them in genetic pathways. Researchers are now learning how the RNAi response can be manipulated, so that expression is enhanced, inhibited or restricted by cell type, intracellular compartment or time. Thus, the lessons learned from these models, particularly the worm, could be used to help researchers tailor RNAi to perform optimally depending on circumstance, which could range from whole genome screening to therapeutic intervention. |
Acknowledgements |
| The authors thank Jonathan Hodgkin for comments on the manuscript. Research in the Kuwabara lab is sponsored by funding from the EU Sixth Research Framework Programme (FP6), a BBSRC studentship (PIJ) and an MRC Senior Non-Clinical Fellowship (PEK). |
Statements of Competing Interests |
| The authors declared no competing interests. |
References
|